Example 2: RSS-BVSR with Various LD Matrices
Xiang Zhu
Last updated: 2024-09-16
Checks: 7 0
Knit directory: rss/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200623) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 941a146. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (rmd/example_2.Rmd) and HTML
(docs/example_2.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | adc0cb7 | Xiang Zhu | 2024-07-03 | Build site. |
| Rmd | f88c14d | Xiang Zhu | 2024-07-03 | wflow_publish("rmd/example_2.Rmd") |
| html | bab3f58 | Xiang Zhu | 2020-06-24 | Build site. |
| html | 9c00612 | Xiang Zhu | 2020-06-23 | Build site. |
| Rmd | e4bd120 | Xiang Zhu | 2020-06-23 | wflow_publish("rmd/example_2.Rmd") |
| html | 91c0f75 | Xiang Zhu | 2020-06-23 | Build site. |
| Rmd | 8fed651 | Xiang Zhu | 2020-06-23 | wflow_publish("rmd/example_2.Rmd") |
Overview
This example illustrates the impact of different LD estimates on the RSS results. Three types of estimated LD matrices are considered: cohort sample LD, panel sample LD and shrinkage panel sample LD in Wen and Stephens, (2010) This example is closely related to Section 4.1 of Zhu and Stephens (2017).
The single-SNP summary-level data are computed from a simulated GWAS dataset. The simulation scheme is described in Section 4.1 Zhu and Stephens (2017). Specifically, 10 “causal” SNPs are randomly drawn from 982 SNPs on chromosome 16, with effect sizes coming from standard normal \({\cal N}(0,1)\). Effect sizes of remaining SNPs are zero. The true PVE (SNP heritability) is 0.2.
Three types of LD estimates are considered here.
cohort sample LD: the sample correlation matrix using genotypes in the cohort (WTCCC UK Blood Service Control Group)
shrinkage panel sample LD: the shrinkage correlation matrix (Wen and Stephens, 2010) using genotypes in the panel (WTCCC 1958 British Birth Cohort)
panel sample LD: the sample correlation matrix using genotypes in the panel (WTCCC 1958 British Birth Cohort)
To reproduce results of Example 2, please read the step-by-step guide
below and run example2.m.
Before running example2.m,
please first install the MCMC
subroutines. Please find installation instructions here.
Step-by-step illustration
Step 1. Download data files.
All data files required to run this example are freely available at
Zenodo ![]() . Please contact me if you have trouble accessing this
file. After a complete download, you should see the following files.
. Please contact me if you have trouble accessing this
file. After a complete download, you should see the following files.
The data file example2.mat contains the following
elements.
betahat: 982 by 1 vector, single-SNP effect size estimate for each SNPse: 982 by 1 vector, standard errors of the single-SNP effect size estimatesNsnp: 982 by 1 vector, sample size of each SNPcohort_R: cohort sample LDshrink_R: shrinkage panel sample LDpanel_R: panel sample LDsnp_info: 3 by 1 cell, ID and allele of each SNP
Step 2. Fit three RSS-BVSR models with different LD matrices.
% cohort sample LD
[betasam, gammasam, hsam, logpisam, Naccept] = rss_bvsr(betahat, se, cohort_R, Nsnp, Ndraw, Nburn, Nthin);
% shrinkage panel sample LD
[betasam, gammasam, hsam, logpisam, Naccept] = rss_bvsr(betahat, se, shrink_R, Nsnp, Ndraw, Nburn, Nthin);
% panel sample LD
[betasam, gammasam, hsam, logpisam, Naccept] = rss_bvsr(betahat, se, panel_R, Nsnp, Ndraw, Nburn, Nthin);More simulations
The simulations in Section 4.1 of Zhu and Stephens
(2017) are essentially “replications” of the example above. The
simulated datasets in Section 4.1 are available as
rss_example2_data_{1/2/3}.tar.gz1.
Each simulated dataset contains three files:
genotype.txt, phenotype.txt and
simulated_data.mat. The files genotype.txt and
phenotype.txt are the genotype and phenotype files for GEMMA. The
file simulated_data.mat contains three cells.
true_para = {pve, beta, gamma, sigma};
individual_data = {y, X};
summary_data = {betahat, se, Nsnp};Only the summary_data cell above is used as the input
for RSS methods.
RSS methods also require an estimated LD matrix. The three types of
LD matrices are provided in the file
genotype2.mat1.
After applying RSS methods to these simulated data, we obtain the following results.
| True PVE = 0.2 |
|---|
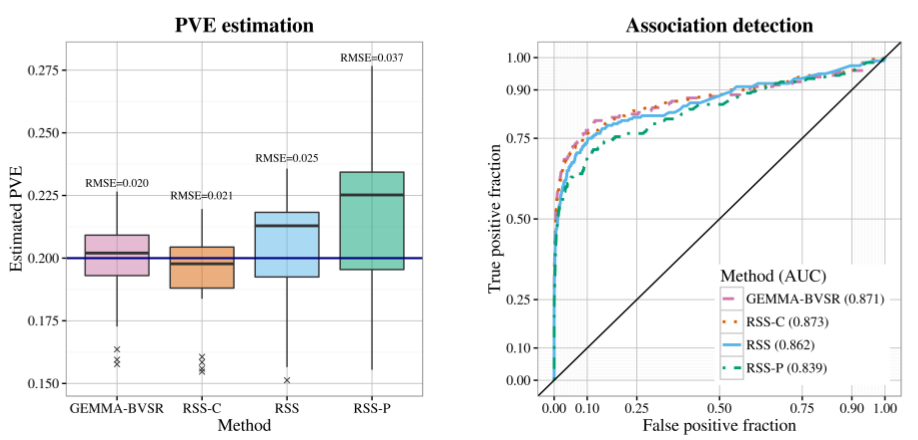 |
| True PVE = 0.02 | True PVE = 0.002 |
|---|---|
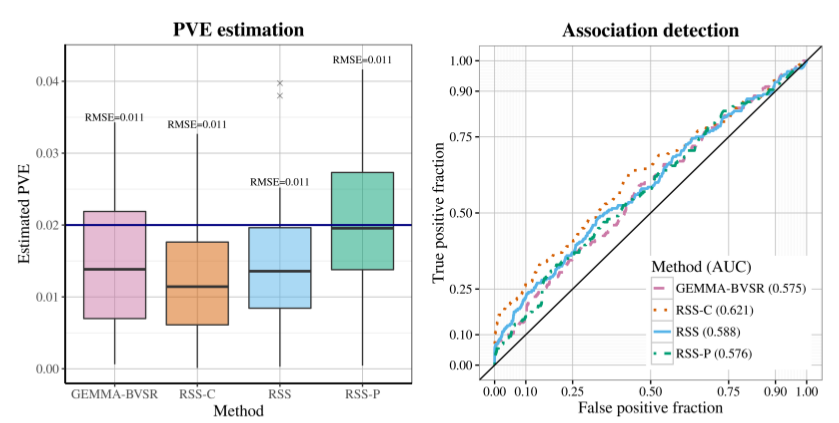 |
 |
Footnotes:
- Currently these files are locked, since they contain individual-level genotypes from Wellcome Trust Case Control Consortium (WTCCC, https://www.wtccc.org.uk/). You need to get permission from WTCCC before we can share these files with you.
─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.4.1 (2024-06-14)
os macOS Sonoma 14.6.1
system aarch64, darwin20
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz America/Los_Angeles
date 2024-09-16
pandoc 3.1.11 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/aarch64/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
bslib 0.8.0 2024-07-29 [1] CRAN (R 4.4.0)
cachem 1.1.0 2024-05-16 [1] CRAN (R 4.4.0)
callr 3.7.6 2024-03-25 [1] CRAN (R 4.4.0)
cli 3.6.3 2024-06-21 [1] CRAN (R 4.4.0)
devtools 2.4.5 2022-10-11 [1] CRAN (R 4.4.0)
digest 0.6.37 2024-08-19 [1] CRAN (R 4.4.1)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.4.0)
evaluate 0.24.0 2024-06-10 [1] CRAN (R 4.4.0)
fansi 1.0.6 2023-12-08 [1] CRAN (R 4.4.0)
fastmap 1.2.0 2024-05-15 [1] CRAN (R 4.4.0)
fs 1.6.4 2024-04-25 [1] CRAN (R 4.4.0)
getPass 0.2-4 2023-12-10 [1] CRAN (R 4.4.0)
git2r 0.33.0 2023-11-26 [1] CRAN (R 4.4.0)
glue 1.7.0 2024-01-09 [1] CRAN (R 4.4.0)
htmltools 0.5.8.1 2024-04-04 [1] CRAN (R 4.4.0)
htmlwidgets 1.6.4 2023-12-06 [1] CRAN (R 4.4.0)
httpuv 1.6.15 2024-03-26 [1] CRAN (R 4.4.0)
httr 1.4.7 2023-08-15 [1] CRAN (R 4.4.0)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.4.0)
jsonlite 1.8.8 2023-12-04 [1] CRAN (R 4.4.0)
knitr 1.48 2024-07-07 [1] CRAN (R 4.4.0)
later 1.3.2 2023-12-06 [1] CRAN (R 4.4.0)
lifecycle 1.0.4 2023-11-07 [1] CRAN (R 4.4.0)
magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.4.0)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.4.0)
mime 0.12 2021-09-28 [1] CRAN (R 4.4.0)
miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.4.0)
pillar 1.9.0 2023-03-22 [1] CRAN (R 4.4.0)
pkgbuild 1.4.4 2024-03-17 [1] CRAN (R 4.4.0)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.4.0)
pkgload 1.4.0 2024-06-28 [1] CRAN (R 4.4.0)
processx 3.8.4 2024-03-16 [1] CRAN (R 4.4.0)
profvis 0.3.8 2023-05-02 [1] CRAN (R 4.4.0)
promises 1.3.0 2024-04-05 [1] CRAN (R 4.4.0)
ps 1.8.0 2024-09-12 [1] CRAN (R 4.4.1)
purrr 1.0.2 2023-08-10 [1] CRAN (R 4.4.0)
R6 2.5.1 2021-08-19 [1] CRAN (R 4.4.0)
Rcpp 1.0.13 2024-07-17 [1] CRAN (R 4.4.0)
remotes 2.5.0 2024-03-17 [1] CRAN (R 4.4.0)
rlang 1.1.4 2024-06-04 [1] CRAN (R 4.4.0)
rmarkdown 2.28 2024-08-17 [1] CRAN (R 4.4.0)
rprojroot 2.0.4 2023-11-05 [1] CRAN (R 4.4.0)
rstudioapi 0.16.0 2024-03-24 [1] CRAN (R 4.4.0)
sass 0.4.9.9000 2024-07-11 [1] Github (rstudio/sass@9228fcf)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.4.0)
shiny 1.9.1 2024-08-01 [1] CRAN (R 4.4.0)
stringi 1.8.4 2024-05-06 [1] CRAN (R 4.4.0)
stringr 1.5.1 2023-11-14 [1] CRAN (R 4.4.0)
tibble 3.2.1 2023-03-20 [1] CRAN (R 4.4.0)
urlchecker 1.0.1 2021-11-30 [1] CRAN (R 4.4.0)
usethis 3.0.0 2024-07-29 [1] CRAN (R 4.4.0)
utf8 1.2.4 2023-10-22 [1] CRAN (R 4.4.0)
vctrs 0.6.5 2023-12-01 [1] CRAN (R 4.4.0)
whisker 0.4.1 2022-12-05 [1] CRAN (R 4.4.0)
workflowr * 1.7.1 2023-08-23 [1] CRAN (R 4.4.0)
xfun 0.47 2024-08-17 [1] CRAN (R 4.4.0)
xtable 1.8-4 2019-04-21 [1] CRAN (R 4.4.0)
yaml 2.3.10 2024-07-26 [1] CRAN (R 4.4.0)
[1] /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/library
──────────────────────────────────────────────────────────────────────────────