Fine-mapping Height on More Regions (SuSiE-RSS with lambda)
Yuxin Zou
10/06/2019
Last updated: 2019-11-17
Checks: 7 0
Knit directory: finemap-uk-biobank/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20191114) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Untracked files:
Untracked: data/height.ACAN.XtX.Xty.rds
Untracked: data/height.ADAMTS10.XtX.Xty.rds
Untracked: data/height.ADAMTS17.XtX.Xty.rds
Untracked: data/height.ADAMTSL3.XtX.Xty.rds
Untracked: data/height.CABLES1.XtX.Xty.rds
Untracked: data/height.CDK6.XtX.Xty.rds
Untracked: data/height.DLEU1.XtX.Xty.rds
Untracked: data/height.EFEMP1.XtX.Xty.rds
Untracked: data/height.GDF5.XtX.Xty.rds
Untracked: data/height.GNA12.XtX.Xty.rds
Untracked: data/height.HHIP.XtX.Xty.rds
Untracked: data/height.HMGA2.XtX.Xty.rds
Untracked: data/height.KDM2A.XtX.Xty.rds
Untracked: data/height.LCORL.XtX.Xty.rds
Untracked: data/height.MTMR11.XtX.Xty.rds
Untracked: data/height.UQCC1.XtX.Xty.rds
Untracked: data/height.ZBTB38.0.01.simulation1000.rds
Untracked: data/height.ZBTB38.XtX.Xty.rds
Untracked: data/height.ZBTB38.neale.rds
Untracked: data/height.ZBTB38.plink2.height.glm.linear
Untracked: data/height.ZBTB38.removeCS1.XtX.Xty.rds
Untracked: data/height.ZBTB38.removeCS2.XtX.Xty.rds
Untracked: data/height.ZBTB38.susie.model.rds
Unstaged changes:
Modified: analysis/_site.yml
Deleted: analysis/finemap_height_more.Rmd
Deleted: analysis/finemap_height_more_rss.Rmd
Deleted: analysis/finemap_height_more_rss_lambda.Rmd
Modified: scripts/plots.R
Modified: scripts/prepare.region.sh
Modified: scripts/prepare.susieinput.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | aa37c36 | zouyuxin | 2019-11-18 | wflow_publish(“analysis/finemap_height_20top_susierss_lambda.Rmd”) |
Load packages
library(dplyr)
library(ggplot2)
library(susieR)
library(data.table)
library(Matrix)
library(gridExtra)
library(readr)
library(cowplot)knitr::read_chunk("scripts/plots.R")library(ggplot2)
#' plot gene name annotations
#' @param dat a matrix of gene names with 'start' and 'end' base-pair position
#' @param xrange range of x axis, base-pair position
plot_geneName = function(dat, xrange, chr){
ngene = 2:nrow(dat)
line = 1
dat$lines = NA
dat$lines[1] = 1
gene.end = dat[1, 'end']
while(length(ngene) != 0){
id = which(dat[ngene, 'start'] > gene.end + 0.02)[1]
if(!is.na(id)){
dat$lines[ngene[id]] = line
gene.end = dat[ngene[id],'end']
ngene = ngene[-id]
}else{
line = line + 1
dat$lines[ngene[1]] = line
gene.end = dat[ngene[1],'end']
ngene = ngene[-1]
}
}
dat$start = pmax(dat$start, xrange[1])
dat$end = pmin(dat$end, xrange[2])
dat$mean = rowMeans(dat[,c('start', 'end')])
pl = ggplot(dat, aes(xmin = xrange[1], xmax = xrange[2])) + xlim(xrange[1], xrange[2]) + ylim(min(-dat$lines-0.6), -0.8) +
geom_rect(aes(xmin = start, xmax = end, ymin = -lines-0.05, ymax = -lines+0.05), fill='blue') +
geom_text(aes(x = mean, y=-lines-0.4, label=geneName), size=4) +
xlab(paste0('base-pair position (Mb) on chromosome ', chr)) + ylab('Gene') +
theme_bw() + theme(axis.text.x=element_blank(),
axis.ticks = element_blank(),
axis.text.y=element_blank(),
axis.title = element_text(size=15),
plot.title=element_text(size=11),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank())
pl
}
discrete_gradient_pal <- function(colours, bins = 5) {
ramp <- scales::colour_ramp(colours)
function(x) {
if (length(x) == 0) return(character())
i <- floor(x * bins)
i <- ifelse(i > bins-1, bins-1, i)
ramp(i/(bins-1))
}
}
scale_colour_discrete_gradient <- function(..., colours, bins = 5, na.value = "grey50", guide = "colourbar", aesthetics = "colour", colors) {
colours <- if (missing(colours))
colors
else colours
continuous_scale(
aesthetics,
"discrete_gradient",
discrete_gradient_pal(colours, bins),
na.value = na.value,
guide = guide,
...
)
}
#' Locuszoom plot
#' @param z a vector of z scores with SNP names
#' @param pos base-pair positions
#' @param gene.pos.map a matrix of gene names with 'start' and 'end' base-pair position
#' @param z.ref.name the reference SNP
#' @param ld correlations between teh reference SNP and the rests
#' @param title title of the plot
#' @param title.size the size of the title
#' @param true the true value
#' @param y.height height of -log10(p) plot and height of the gene name annotation plot
#' @param y.lim range of y axis
locus.zoom = function(z, pos, chr, gene.pos.map=NULL, z.ref.name=NULL, ld=NULL,
title = NULL, title.size = 10, true = NULL,
y.height=c(5,1.5), y.lim=NULL, y.type='logp',xrange=NULL){
if(is.null(xrange)){
xrange = c(min(pos), max(pos))
}
tmp = data.frame(POS = pos, p = -(pnorm(-abs(z), log.p = T) + log(2))/log(10), z = z)
if(!is.null(ld) && !is.null(z.ref.name)){
tmp$ref = names(z) == z.ref.name
tmp$r2 = ld^2
if(y.type == 'logp'){
pl_zoom = ggplot(tmp, aes(x = POS, y = p, shape = ref, size=ref, color=r2)) + geom_point() +
ylab("-log10(p value)") + ggtitle(title) + xlim(xrange[1], xrange[2]) +
scale_color_gradientn(colors = c("darkblue", "deepskyblue", "lightgreen", "orange", "red"),
values = seq(0,1,0.2), breaks=seq(0,1,0.2)) +
# scale_colour_discrete_gradient(
# colours = c("darkblue", "deepskyblue", "lightgreen", "orange", "red"),
# limits = c(0, 1.01),
# breaks = c(0,0.2,0.4,0.6,0.8,1),
# guide = guide_colourbar(nbin = 100, raster = FALSE, frame.colour = "black", ticks.colour = NA)
# ) +
scale_shape_manual(values=c(20, 18), guide=FALSE) + scale_size_manual(values=c(2,5), guide=FALSE) +
theme_bw() + theme(axis.title.x=element_blank(),
axis.title.y = element_text(size=15),axis.text = element_text(size=15),
plot.title = element_text(size=title.size))
}else if(y.type == 'z'){
pl_zoom = ggplot(tmp, aes(x = POS, y = z, shape = ref, size=ref, color=r2)) + geom_point() +
ylab("z score") + ggtitle(title) + xlim(xrange[1], xrange[2]) +
scale_color_gradientn(colors = c("darkblue", "deepskyblue", "lightgreen", "orange", "red"),
values = seq(0,1,0.2), breaks=seq(0,1,0.2)) +
# scale_colour_discrete_gradient(
# colours = c("darkblue", "deepskyblue", "lightgreen", "orange", "red"),
# limits = c(0, 1.01),
# breaks = c(0,0.2,0.4,0.6,0.8,1),
# guide = guide_colourbar(nbin = 100, raster = FALSE, frame.colour = "black", ticks.colour = NA)
# ) +
scale_shape_manual(values=c(20, 18), guide=FALSE) + scale_size_manual(values=c(2,5), guide=FALSE) +
theme_bw() + theme(axis.title.x=element_blank(),
axis.title.y = element_text(size=15),axis.text = element_text(size=15),
plot.title = element_text(size=title.size))
}
}else{
if(y.type == 'logp'){
pl_zoom = ggplot(tmp, aes(x = POS, y = p)) + geom_point(color = 'darkblue') +
ylab("-log10(p value)") + ggtitle(title) + xlim(xrange[1], xrange[2]) +
theme_bw() + theme(axis.title.x=element_blank(),
plot.title = element_text(size=title.size))
}else if(y.type == 'z'){
pl_zoom = ggplot(tmp, aes(x = POS, y = z)) + geom_point(color = 'darkblue') +
ylab("z scores") + ggtitle(title) + xlim(xrange[1], xrange[2]) +
theme_bw() + theme(axis.title.x=element_blank(),
plot.title = element_text(size=title.size))
}
}
if(!is.null(y.lim)){
pl_zoom = pl_zoom + ylim(y.lim[1], y.lim[2])
}
# pl_zoom = pl_zoom + geom_hline(yintercept=-log10(5e-08), linetype='dashed', color = 'red')
if(!is.null(true)){
tmp.true = data.frame(POS = which(true!=0), p = tmp$p[which(true!=0)],
ref = (names(z) == z.ref.name)[which(true!=0)],
label = paste0('SNP',1:length(which(true!=0))))
pl_zoom = pl_zoom + geom_point(data=tmp.true, aes(x=POS, y=p),
color='red', show.legend = FALSE, shape=1, stroke = 1) +
geom_text(data=tmp.true, aes(x = POS-30, y=p+1, label=label), size=3, color='red')
}
if(!is.null(gene.pos.map)){
pl_gene = plot_geneName(gene.pos.map, xrange = xrange, chr=chr)
g = egg::ggarrange(pl_zoom, pl_gene, nrow=2, heights = y.height, draw=FALSE)
}else{
g = pl_zoom
}
g
}
#' SuSiE plot with Locuszoom plot
#' @param z a vector of z scores with SNP names
#' @param model the fitted SuSiE model
#' @param pos base-pair positions
#' @param gene.pos.map a matrix of gene names with 'start' and 'end' base-pair position
#' @param z.ref.name the reference SNP
#' @param ld correlations between teh reference SNP and the rests
#' @param title title of the plot
#' @param title.size the size of the title
#' @param true the true value
#' @param plot.locuszoom whether to plot locuszoom plot
#' @param y.lim range of y axis
#' @param y.susie the y axis of the SuSiE plot, 'PIP' or 'p' or 'z', 'p' refers to -log10(p)
susie_plot_locuszoom = function(z, model, pos, chr, gene.pos.map = NULL, z.ref.name, ld,
title = NULL, title.size = 10, true = NULL,
plot.locuszoom = TRUE, y.lim=NULL, y.susie='PIP', xrange=NULL){
if(is.null(xrange)){
xrange = c(min(pos), max(pos))
}
if(plot.locuszoom){
if(y.susie == 'z'){
y.type = 'z'
}else{
y.type = 'logp'
}
pl_zoom = locus.zoom(z, pos = pos, chr = chr, ld=ld, z.ref.name = z.ref.name, title = title, title.size = title.size, y.lim=y.lim, y.type=y.type, xrange=xrange)
}
pip = model$pip
tmp = data.frame(POS = pos, PIP = pip, p = -(pnorm(-abs(z), log.p = T) + log(2))/log(10), z = z)
if(y.susie == 'PIP'){
pl_susie = ggplot(tmp, aes(x = POS, y = PIP)) + geom_point(show.legend = FALSE, size=3) +
xlim(xrange[1], xrange[2]) +
theme_bw() + theme(axis.title.x=element_blank(), axis.text.x=element_blank(),axis.text = element_text(size=15),
axis.title.y = element_text(size=15))
if(!plot.locuszoom){
pl_susie = pl_susie + ggtitle(title) + theme(plot.title = element_text(size=title.size))
}
}else if(y.susie == 'p'){
pl_susie = ggplot(tmp, aes(x = POS, y = p)) + geom_point(show.legend = FALSE, size=3) +
ylab("-log10(p value)") + xlim(xrange[1], xrange[2]) +
theme_bw() + theme(axis.title.x=element_blank(), axis.text.x=element_blank(),axis.text = element_text(size=15),
axis.title.y = element_text(size=15))
if(!plot.locuszoom){
pl_susie = pl_susie + ggtitle(title) + theme(plot.title = element_text(size=title.size))
# pl_susie = pl_susie + geom_hline(yintercept=-log10(5e-08), linetype='dashed', color = 'red')
}
}else if(y.susie == 'z'){
pl_susie = ggplot(tmp, aes(x = POS, y = z)) + geom_point(show.legend = FALSE, size=3) +
ylab("z scores") + xlim(xrange[1], xrange[2]) +
theme_bw() + theme(axis.title.x=element_blank(), axis.text.x=element_blank(),axis.text = element_text(size=15),
axis.title.y = element_text(size=15))
if(!plot.locuszoom){
pl_susie = pl_susie + ggtitle(title) + theme(plot.title = element_text(size=title.size))
}
}
if(!is.null(true)){
tmp.true = data.frame(POS = pos[which(true!=0)], PIP = pip[which(true!=0)])
pl_susie = pl_susie + geom_point(data=tmp.true, aes(x=POS, y=PIP),
color='red', size=3, show.legend = FALSE)
}
model.cs = model$sets$cs
if(!is.null(model.cs)){
tmp$CS = numeric(length(z))
for(i in 1:length(model.cs)){
tmp$CS[model.cs[[i]]] = gsub('L', 'CS', names(model.cs)[i])
}
tmp.cs = tmp[unlist(model.cs),]
tmp.cs$CS = factor(tmp.cs$CS)
levels(tmp.cs$CS) = paste0('CS', 1:length(model.cs))
colors = c('red', 'cyan', 'green', 'orange', 'dodgerblue', 'violet', 'gold',
'#FF00FF', 'forestgreen', '#7A68A6')
if(y.susie == 'PIP'){
pl_susie = pl_susie + geom_point(data=tmp.cs, aes(x=POS, y=PIP, color=CS),
size=3, shape=1, stroke = 2) +
scale_color_manual(values=colors)
}else if(y.susie == 'p'){
pl_susie = pl_susie + geom_point(data=tmp.cs, aes(x=POS, y=p, color=CS),
shape=1, size=3, stroke=1.5) +
scale_color_manual(values=colors)
}else if(y.susie == 'z'){
pl_susie = pl_susie + geom_point(data=tmp.cs, aes(x=POS, y=z, color=CS),
shape=1, size=3, stroke=1.5) +
scale_color_manual(values=colors)
}
}
if(!is.null(gene.pos.map)){
pl_gene = plot_geneName(gene.pos.map, xrange = xrange, chr=chr)
if(plot.locuszoom){
g = egg::ggarrange(pl_zoom, pl_susie, pl_gene, nrow=3, heights = c(4,4,1.5), draw=FALSE)
}else{
g = egg::ggarrange(pl_susie, pl_gene, nrow=2, heights = c(5.5,1.5), draw=FALSE)
}
}else{
if(plot.locuszoom){
g = egg::ggarrange(pl_zoom, pl_susie, nrow=2, heights = c(4,4), draw=FALSE)
}else{
g = pl_susie
}
}
g
}We extract the 16 GWAS regions chosen from GWAS for height. We select 20 top hits and exclude 4 hits on chr6. The region is +- 500kb around the top hit. We exclude SNPs with MAF < 0.01, or inputation score less than 0.9.
We choose this region based on result from http://big.stats.ox.ac.uk/pheno/biobank_50.
| Region | CHR | FROM (Mb) | TO (Mb) |
|---|---|---|---|
| GDF5 | 20 | 33.525756 | 34.525756 |
| ZBTB38 | 3 | 140.621814 | 141.621814 |
| HHIP | 4 | 145.066477 | 146.066477 |
| LCORL | 4 | 17.497066 | 18.497066 |
| EFEMP1 | 2 | 55.606928 | 56.606928 |
| KDM2A | 11 | 66.524534 | 67.524534 |
| HMGA2 | 12 | 65.871880 | 66.871880 |
| CABLES1 | 18 | 20.224810 | 21.224810 |
| ADAMTS10 | 19 | 8.170147 | 9.170147 |
| GNA12 | 7 | 2.302522 | 3.302522 |
| ADAMTS17 | 15 | 100.192953 | 101.192953 |
| CDK6 | 7 | 91.750140 | 92.750140 |
| MTMR11 | 1 | 149.406413 | 150.406413 |
| ADAMTSL3 | 15 | 84.080582 | 85.080582 |
| DLEU1 | 13 | 50.606788 | 51.606788 |
| ACAN | 15 | 88.897827 | 89.897827 |
The code to get phenotype height is get_height. The code to compute summary statistics is in prepare.
Get gene data
genes <- read_delim("data/seq_gene.md.gz",delim = "\t",quote = "")
class(genes) <- "data.frame"
genes <- subset(genes,
group_label == "GRCh37.p5-Primary Assembly" &
feature_type == "GENE")GDF5
Get summary statistics:
region.name = 'GDF5'
file = dir("data/", pattern=paste0("height.",region.name))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2331 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)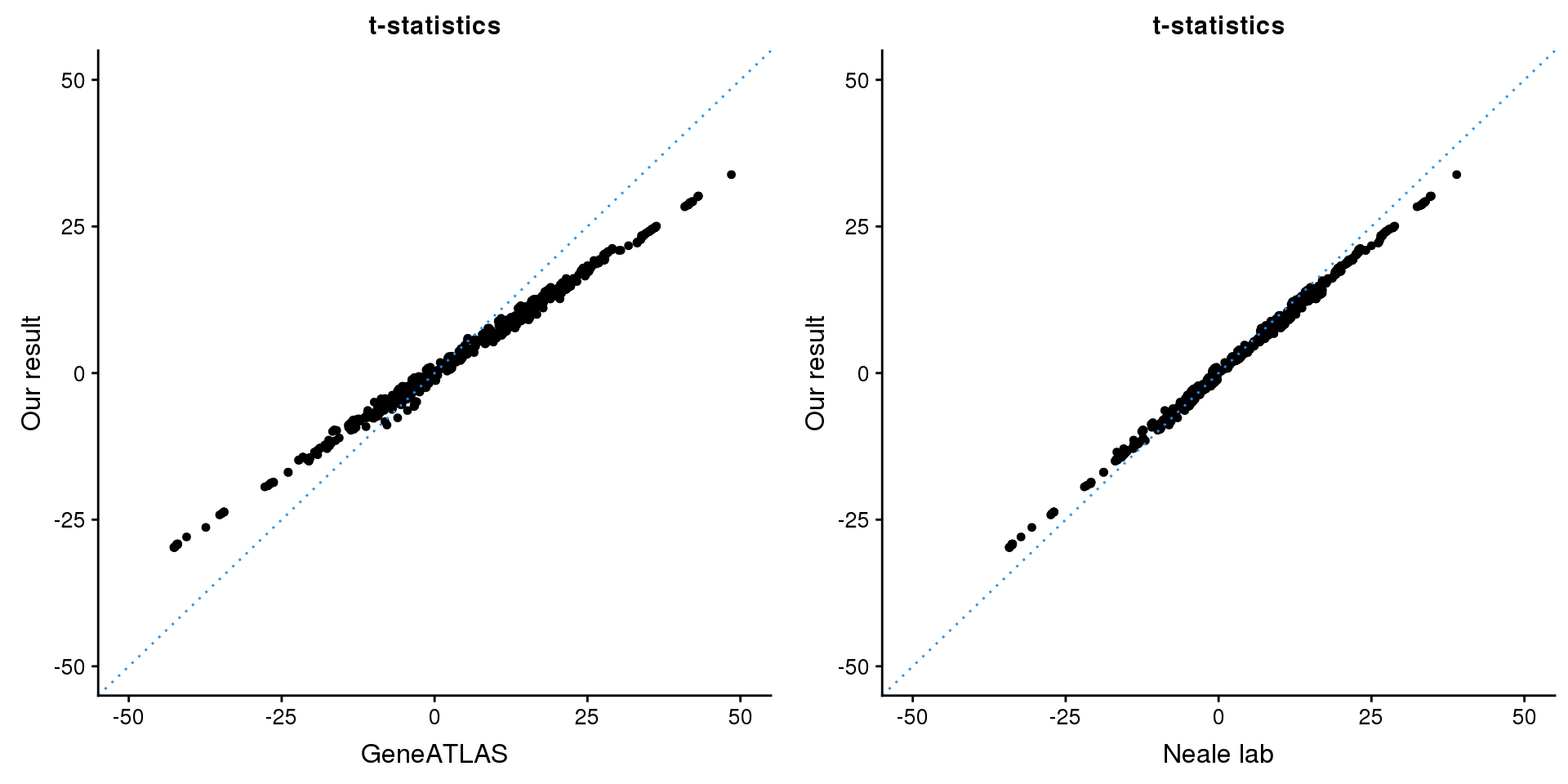
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)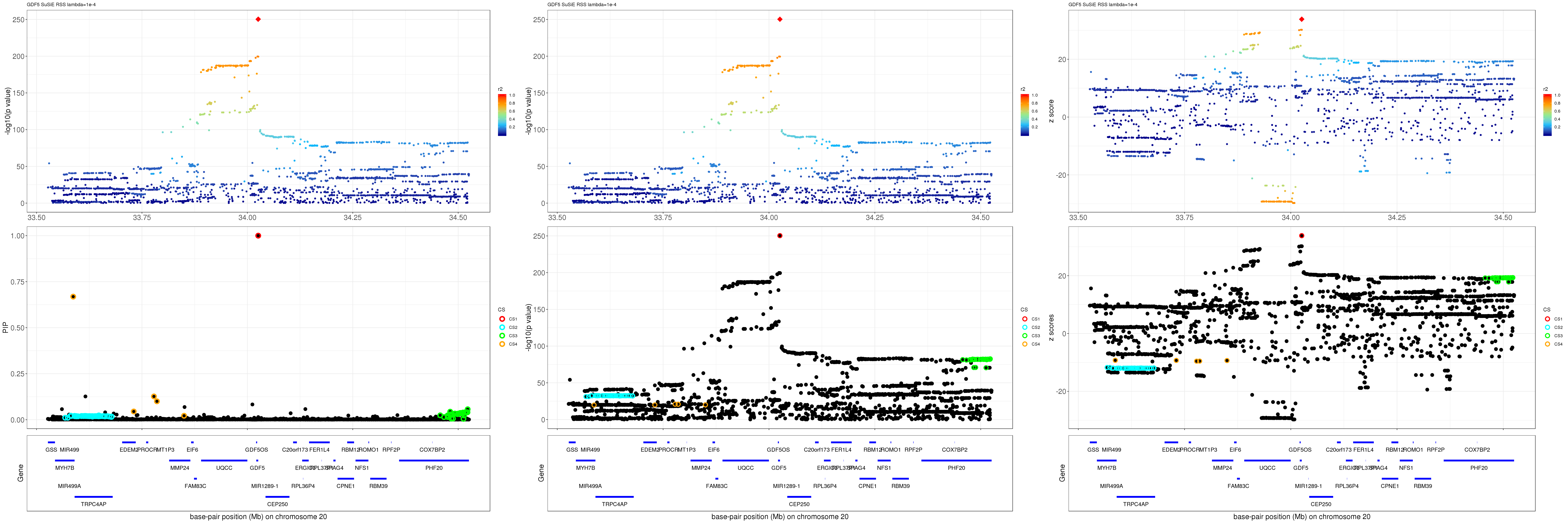
ZBTB38
Get summary statistics:
region.name = 'ZBTB38'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2701 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)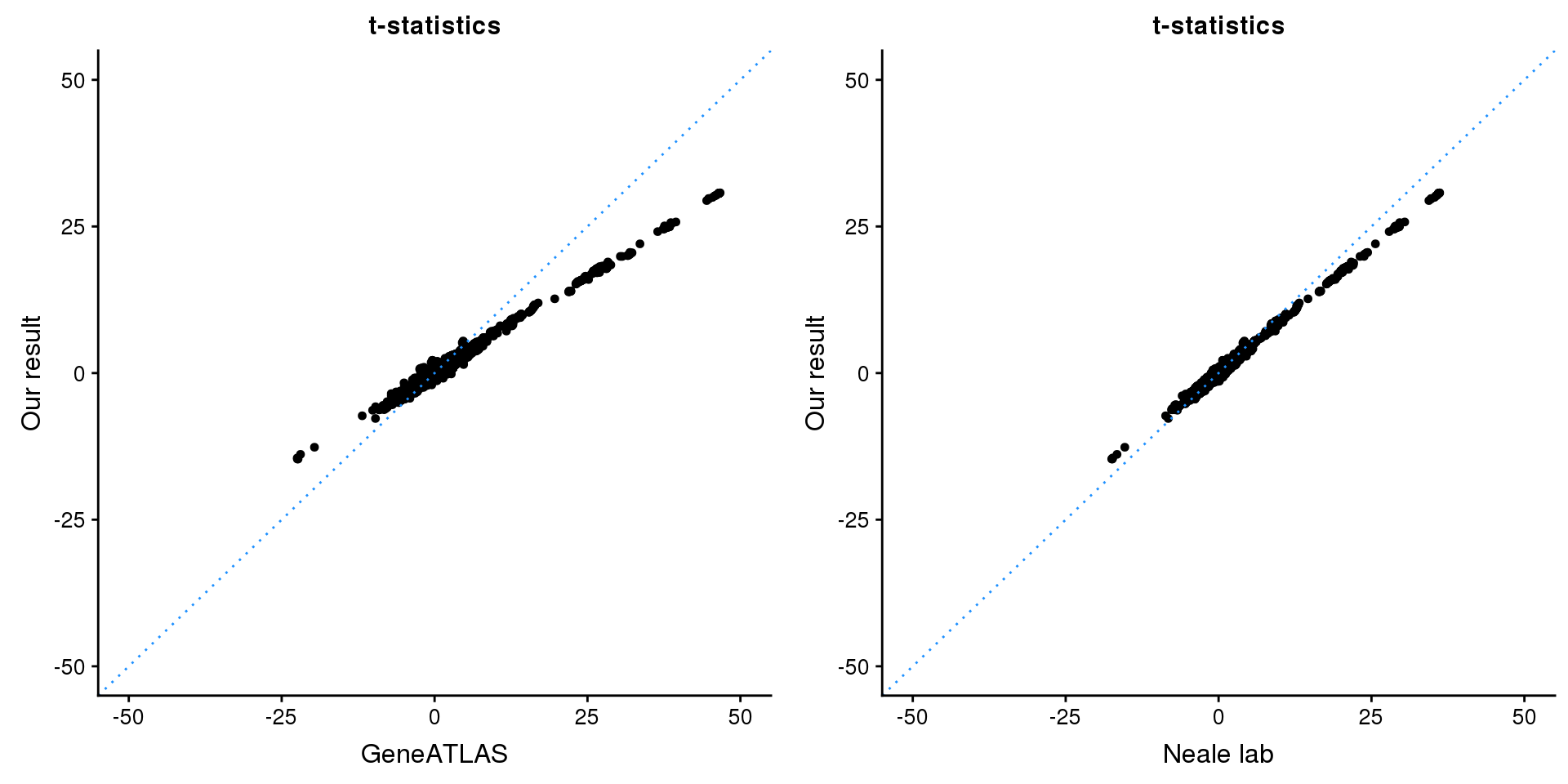
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(5,10,11,16),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)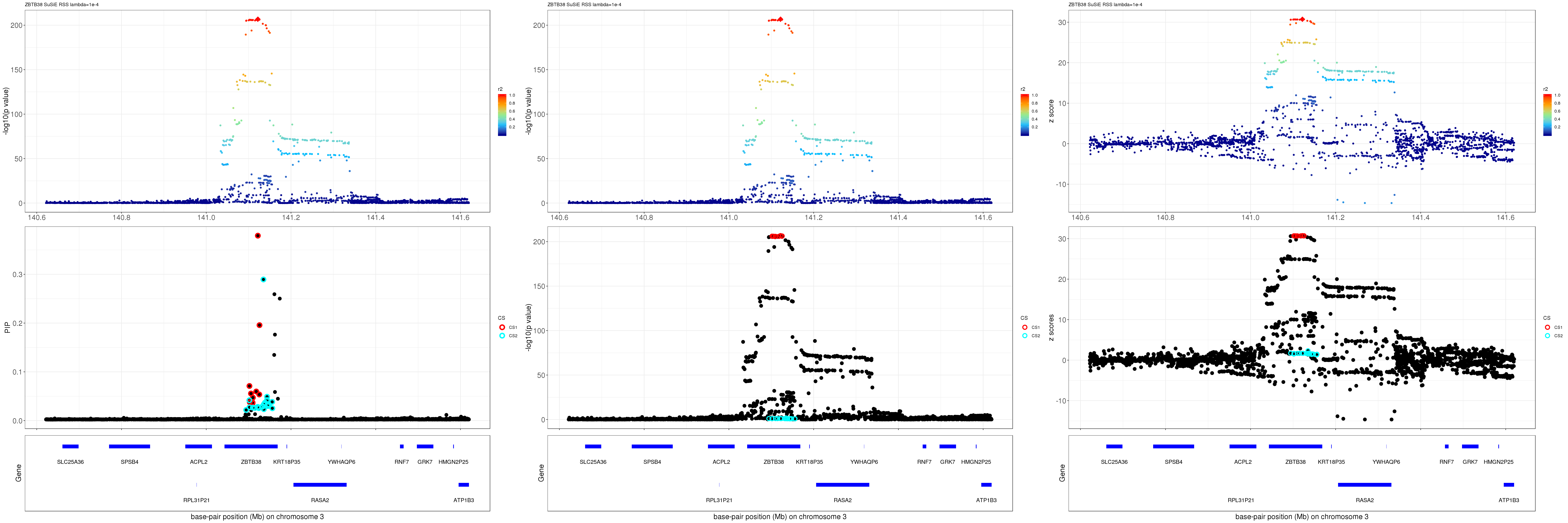
HHIP
Get summary statistics:
region.name = 'HHIP'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "1743 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)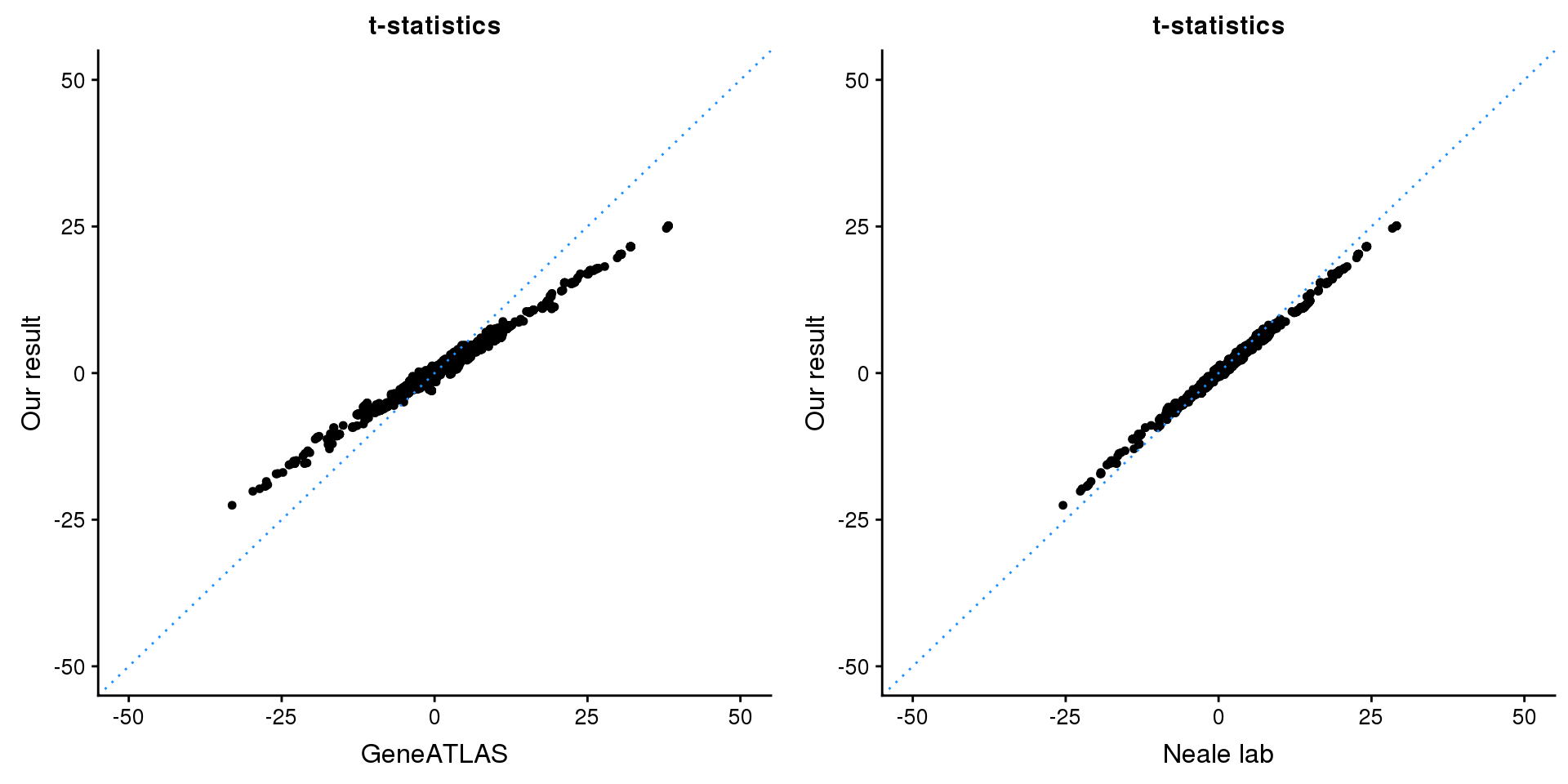
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(2, 4),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)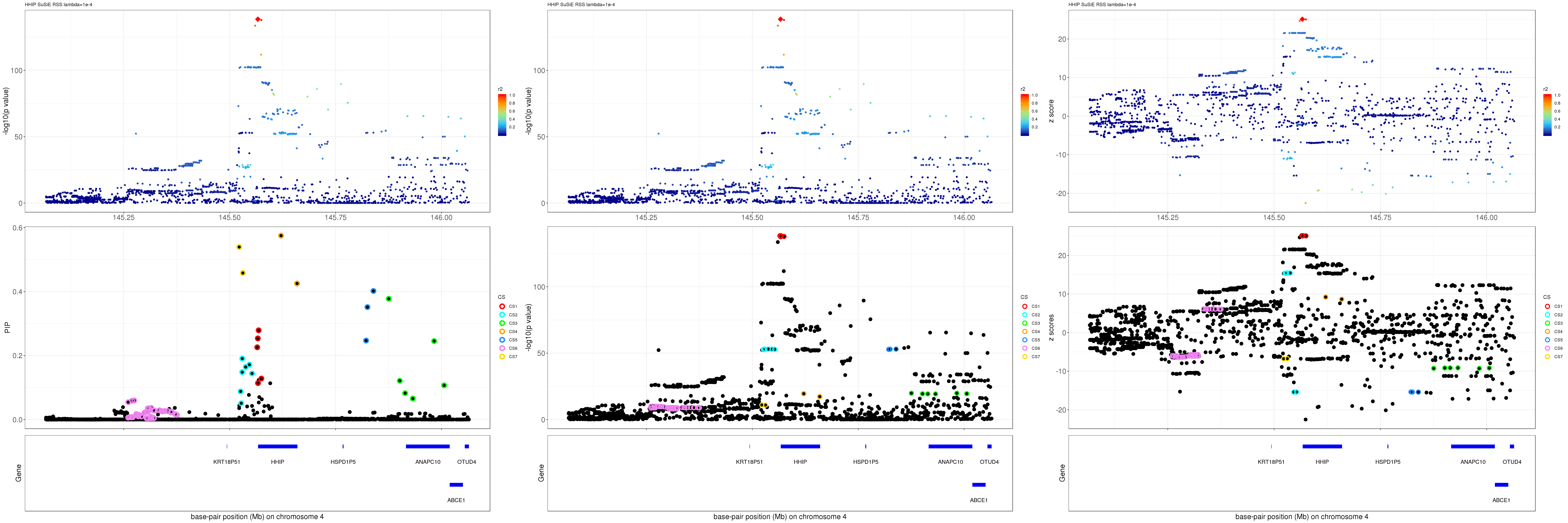
LCORL
Get summary statistics:
region.name = 'LCORL'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2846 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)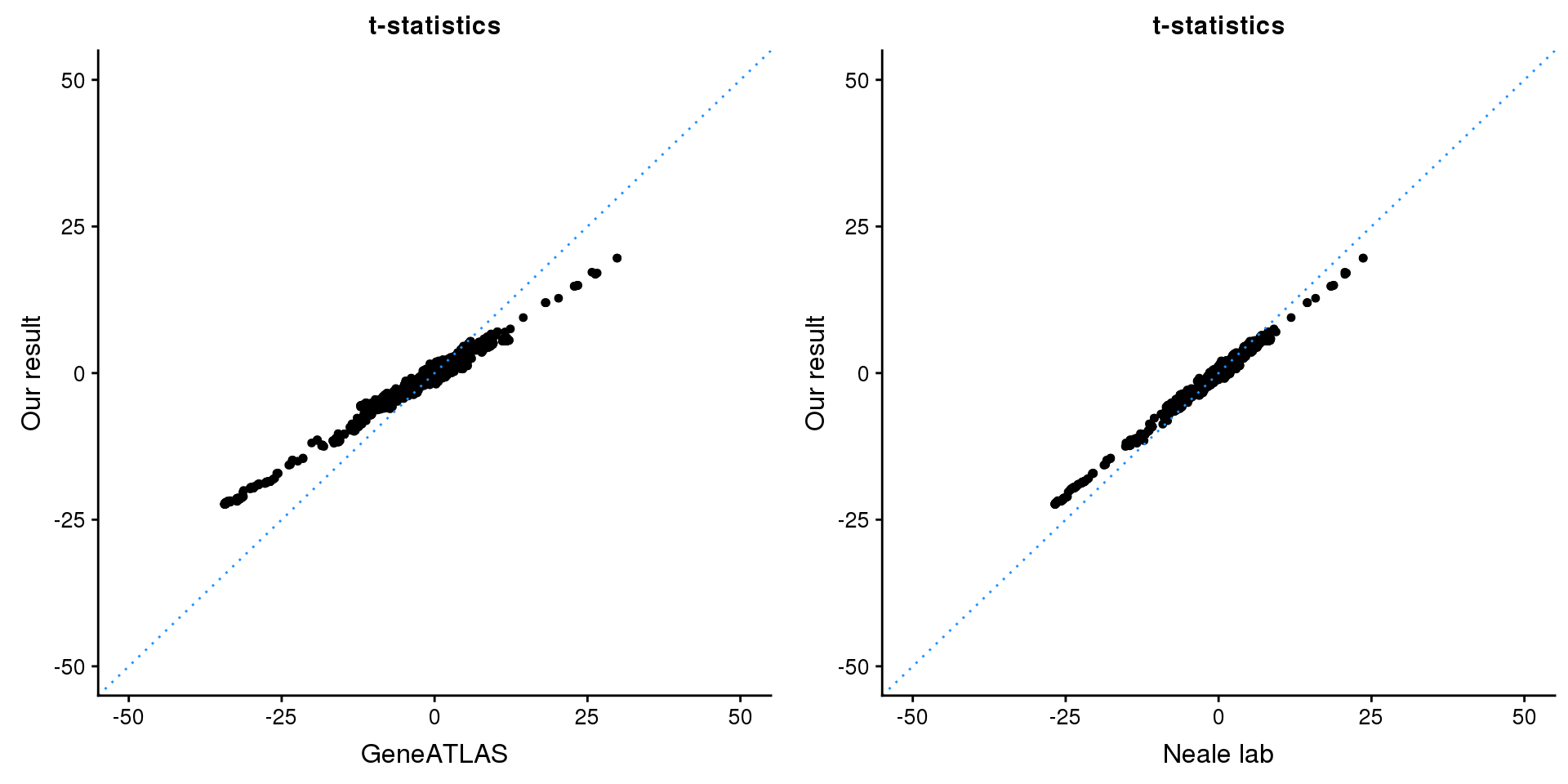
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(3, 10),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)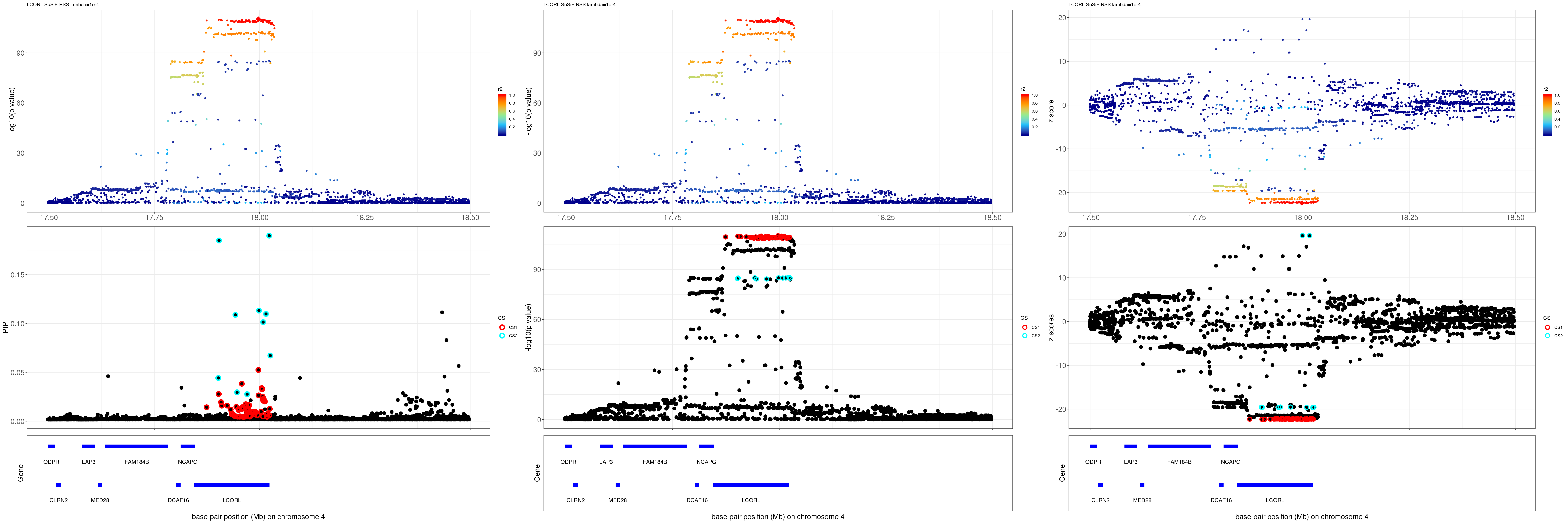
EFEMP1
Get summary statistics:
region.name = 'EFEMP1'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "3710 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)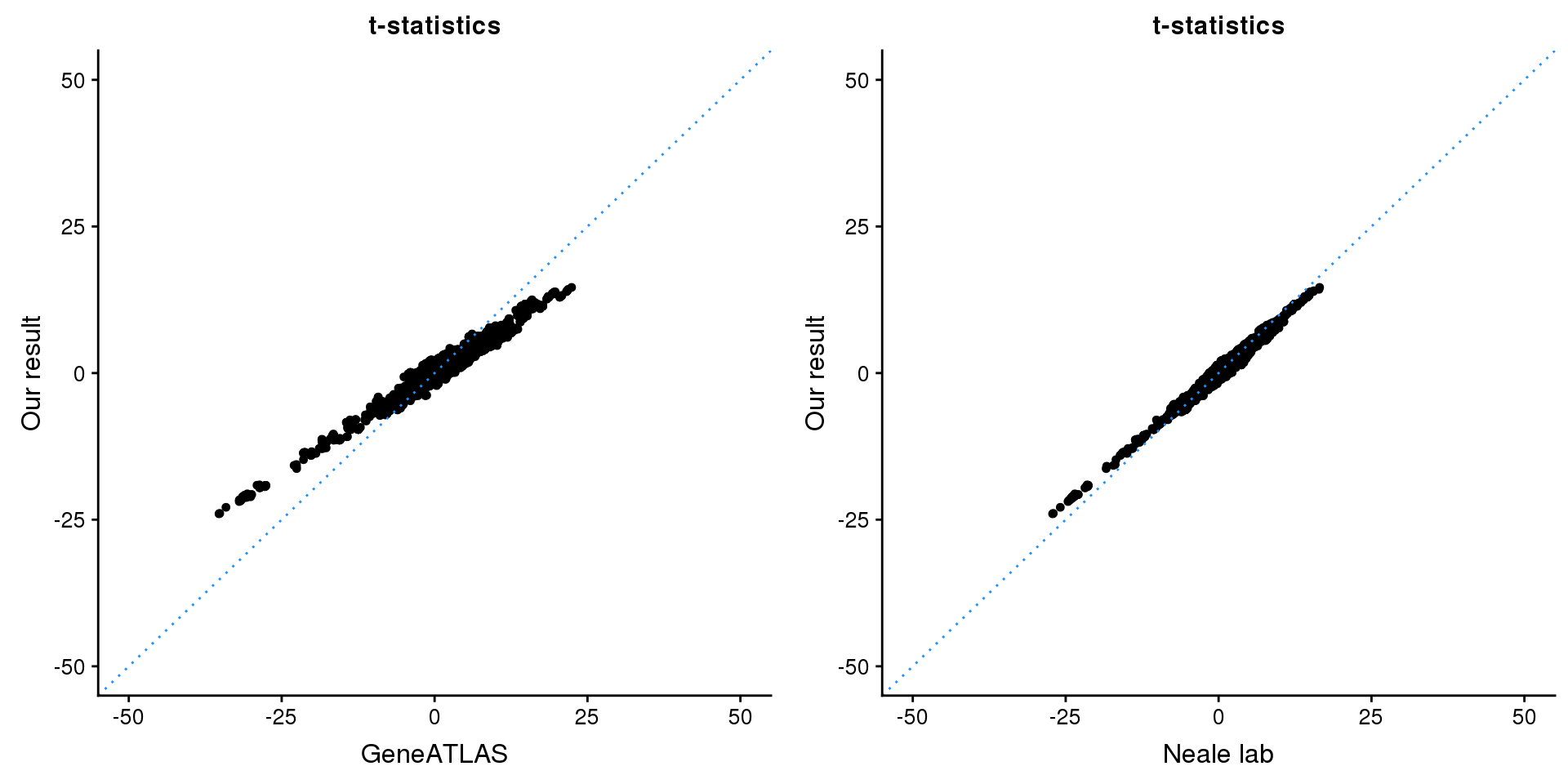
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(10),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)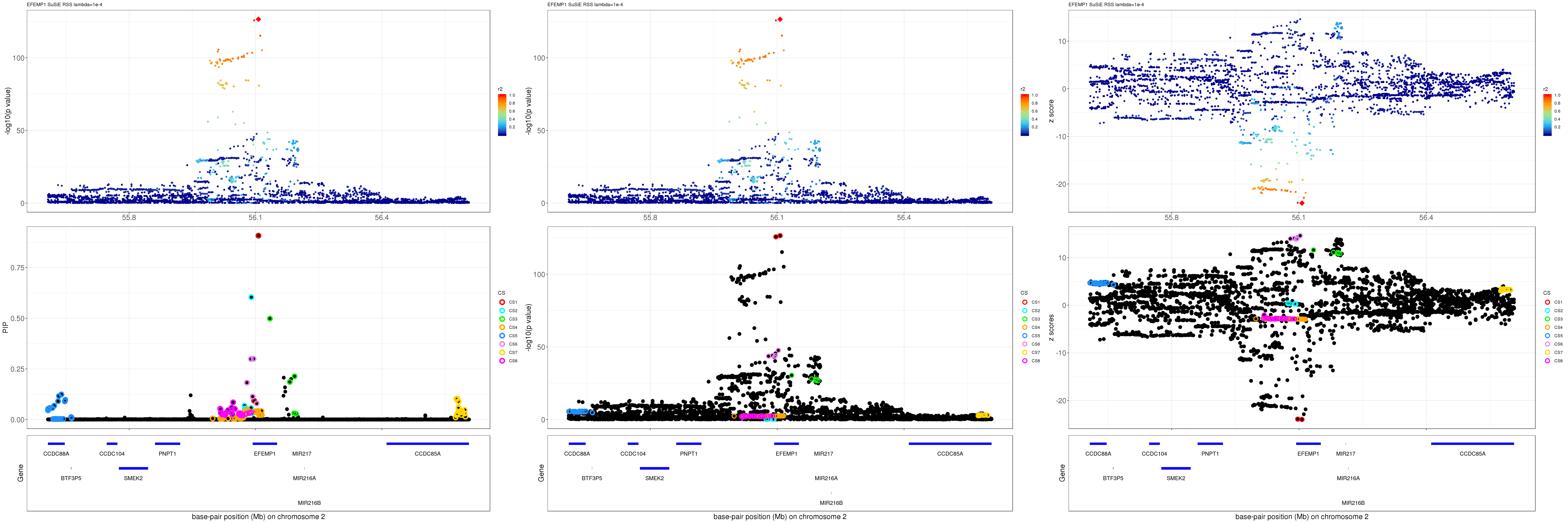
KDM2A
Get summary statistics:
region.name = 'KDM2A'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "1758 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)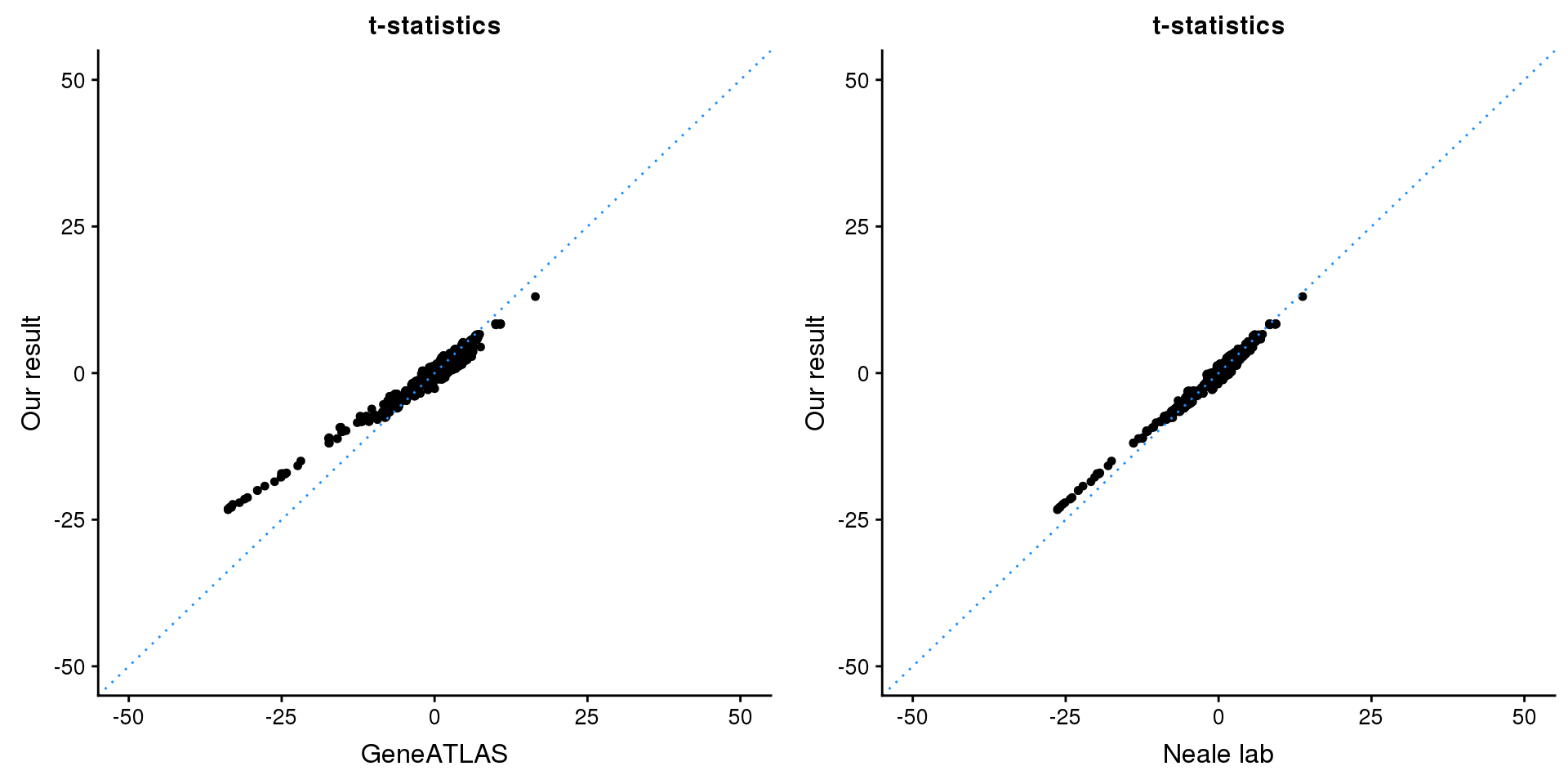
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(2, 16, 44, 45),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)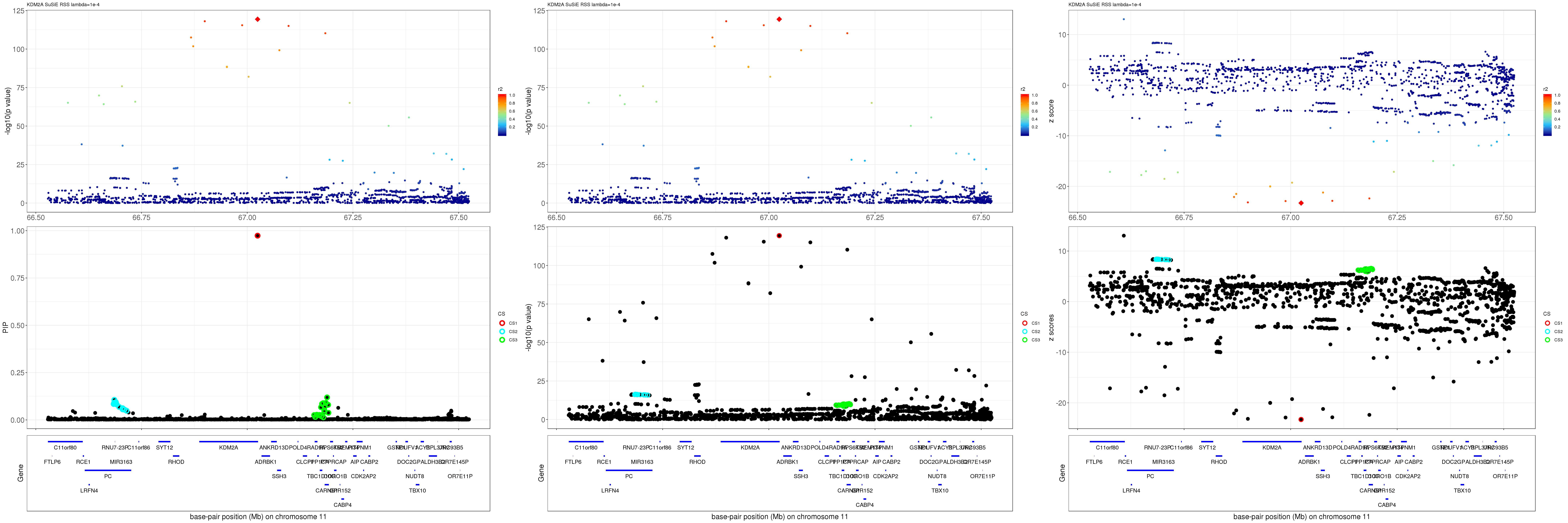
HMGA2
Get summary statistics:
region.name = 'HMGA2'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2109 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)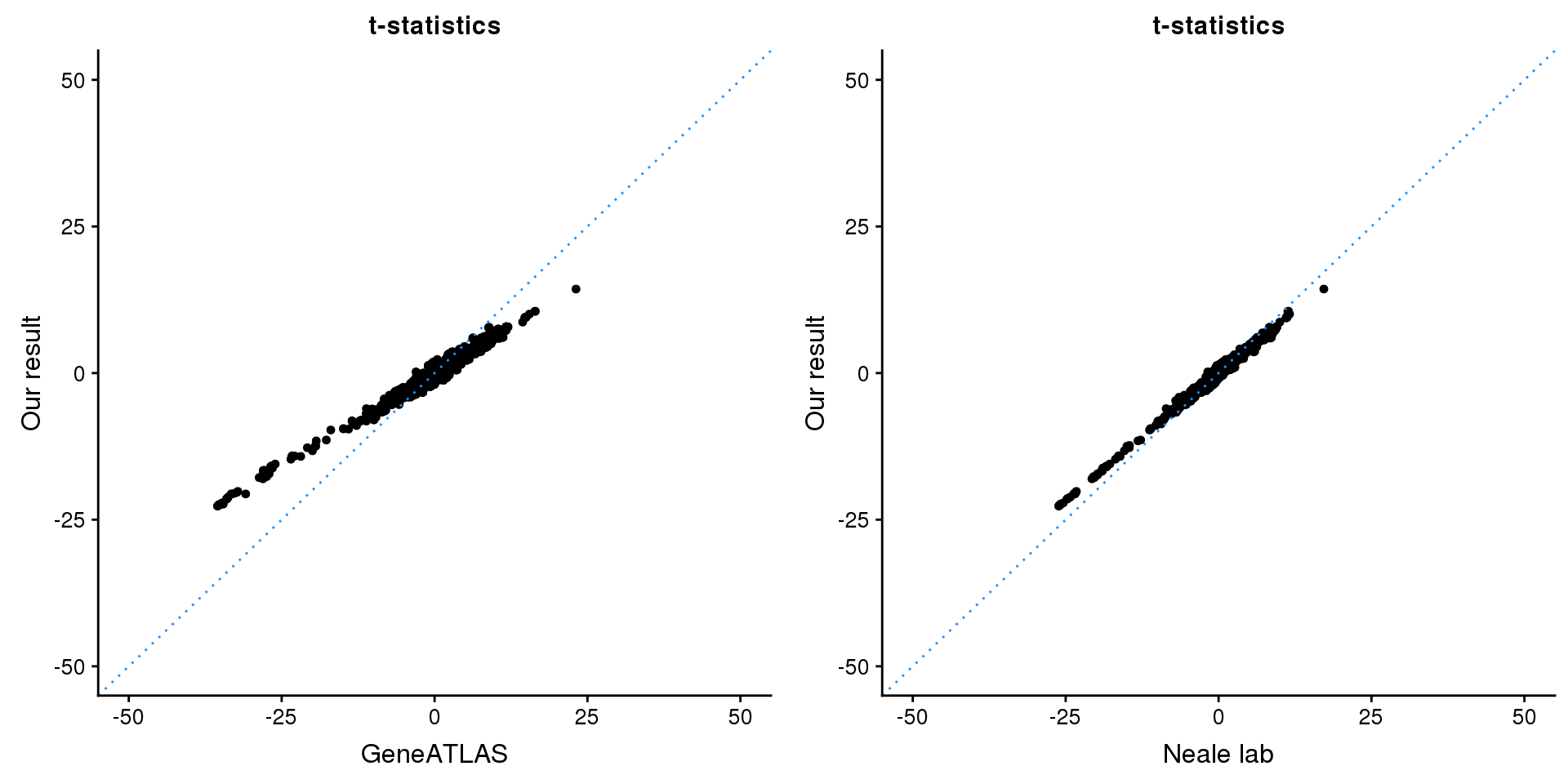
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(1, 5, 11),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)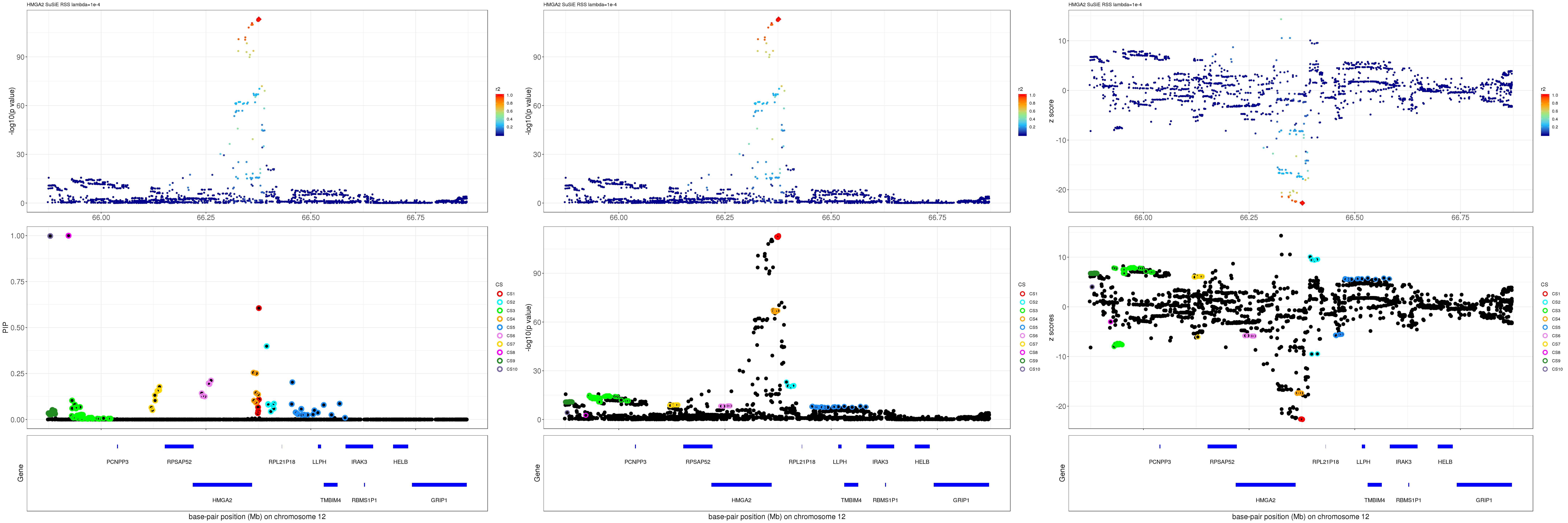
CABLES1
Get summary statistics:
region.name = 'CABLES1'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2166 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)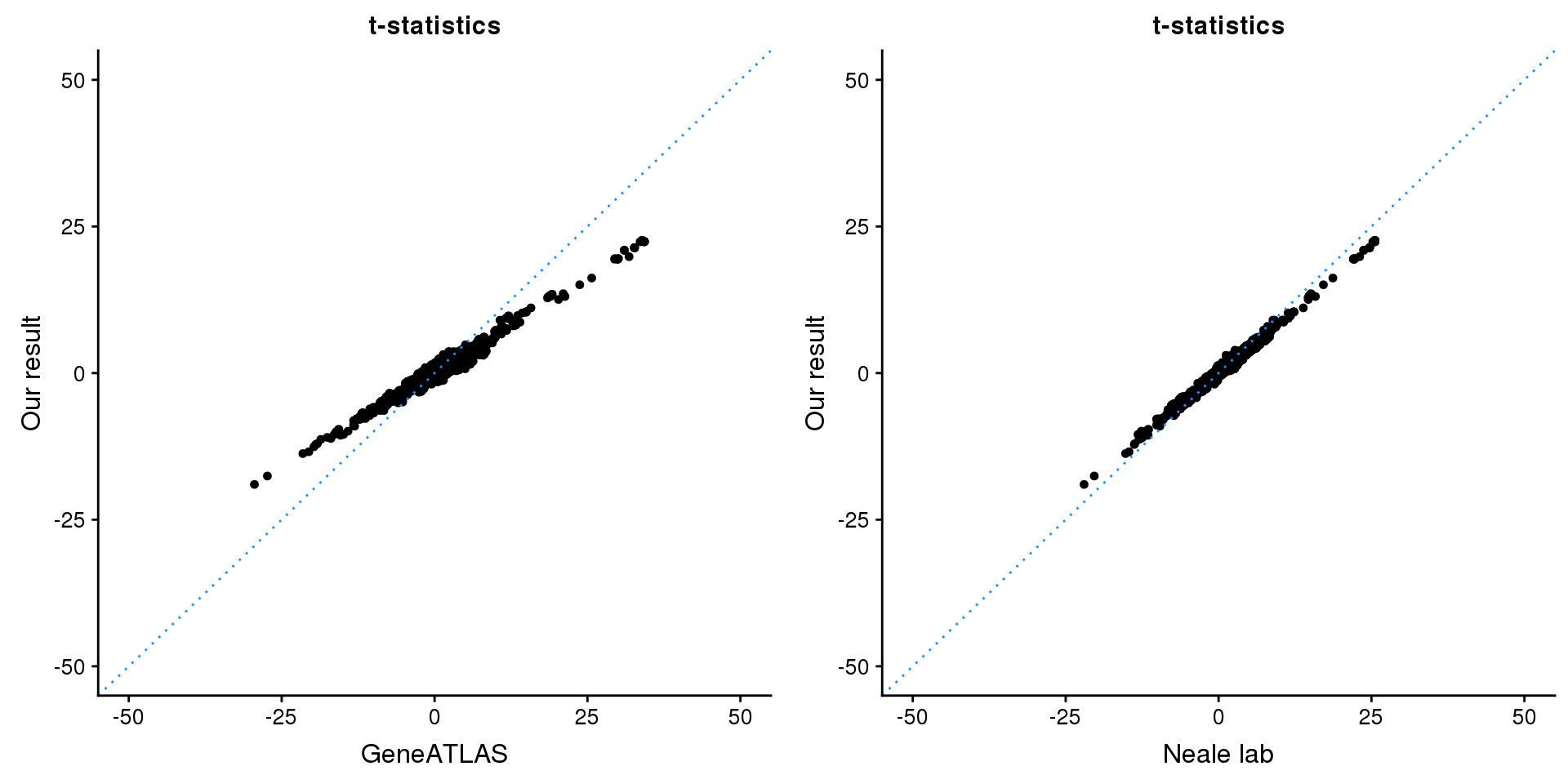
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(1),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)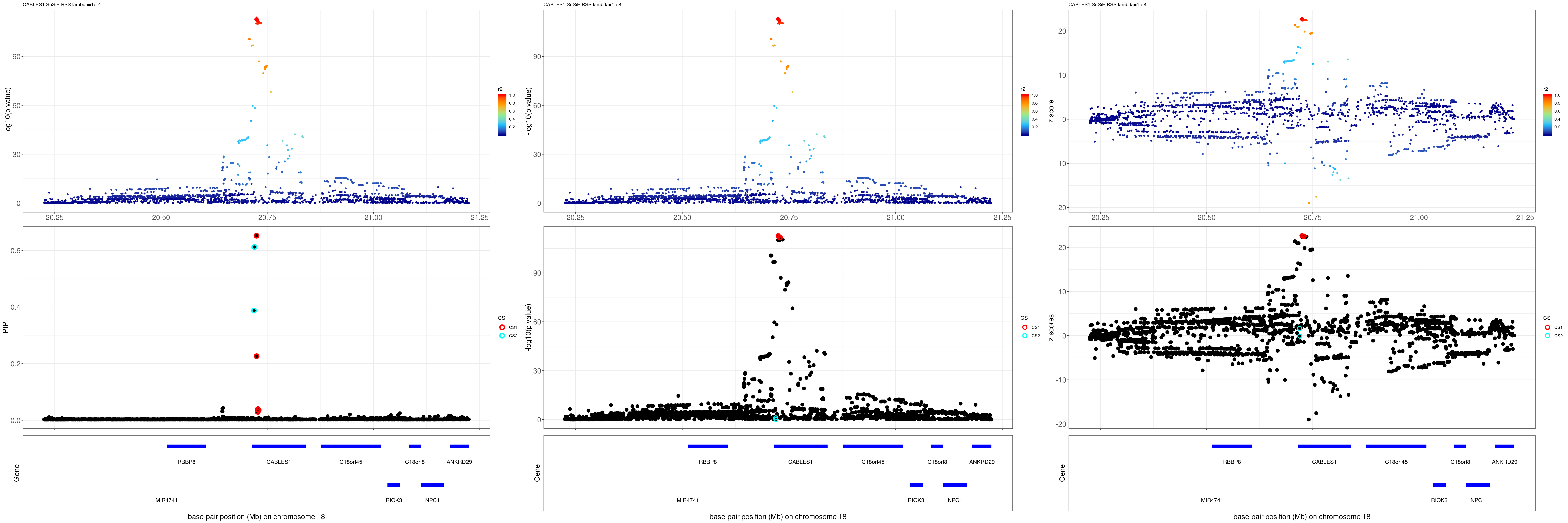
ADAMTS10
Get summary statistics:
region.name = 'ADAMTS10'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "3161 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)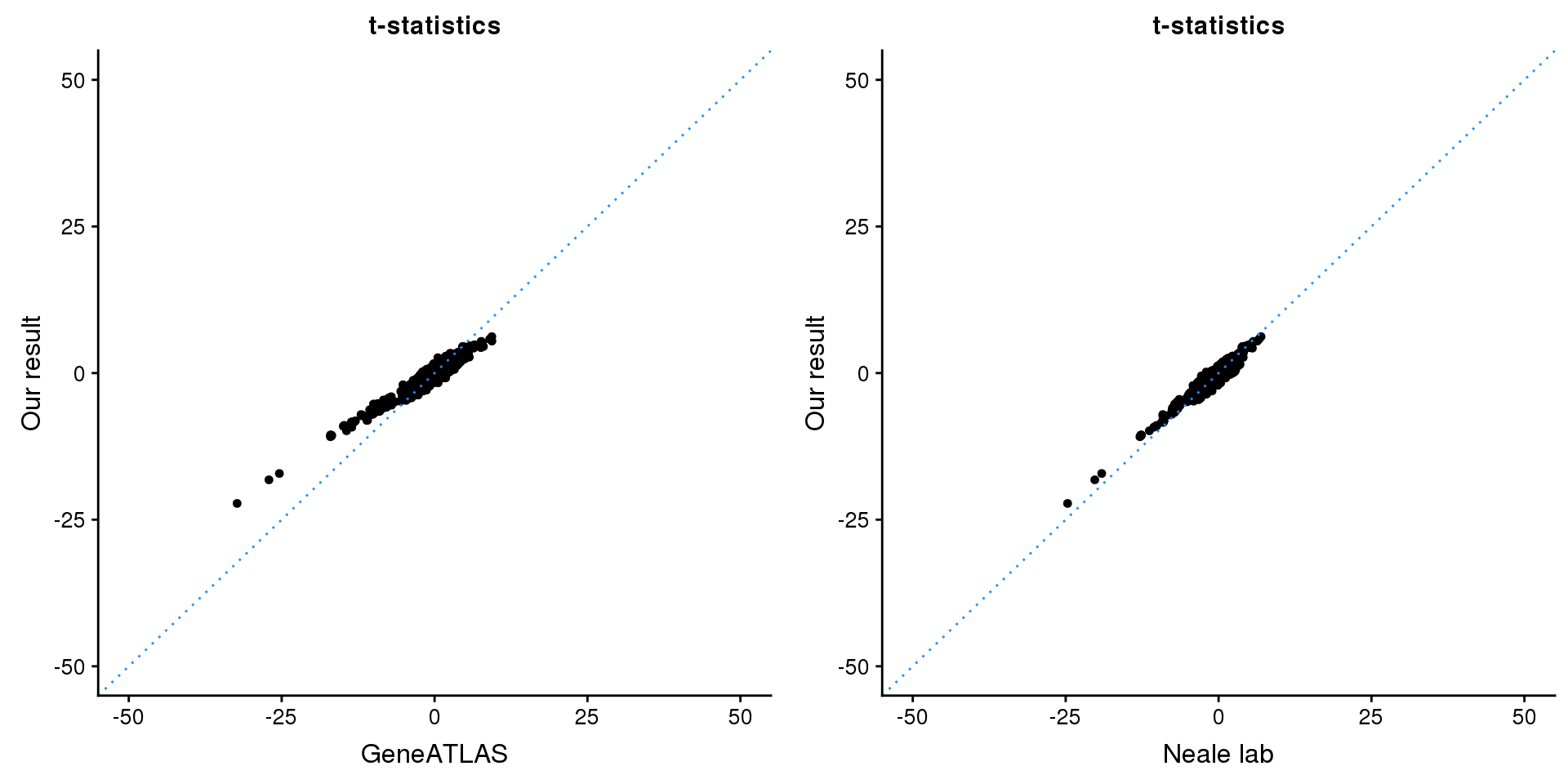
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(2,8,10,24),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)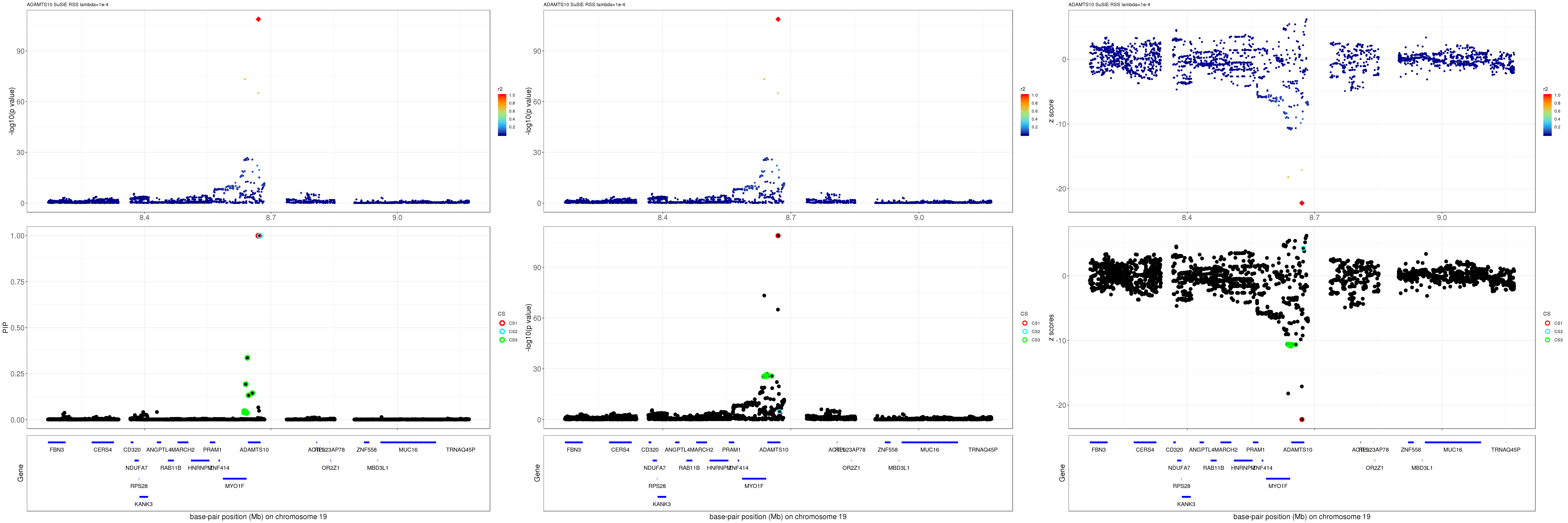
GNA12
Get summary statistics:
region.name = 'GNA12'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "4206 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)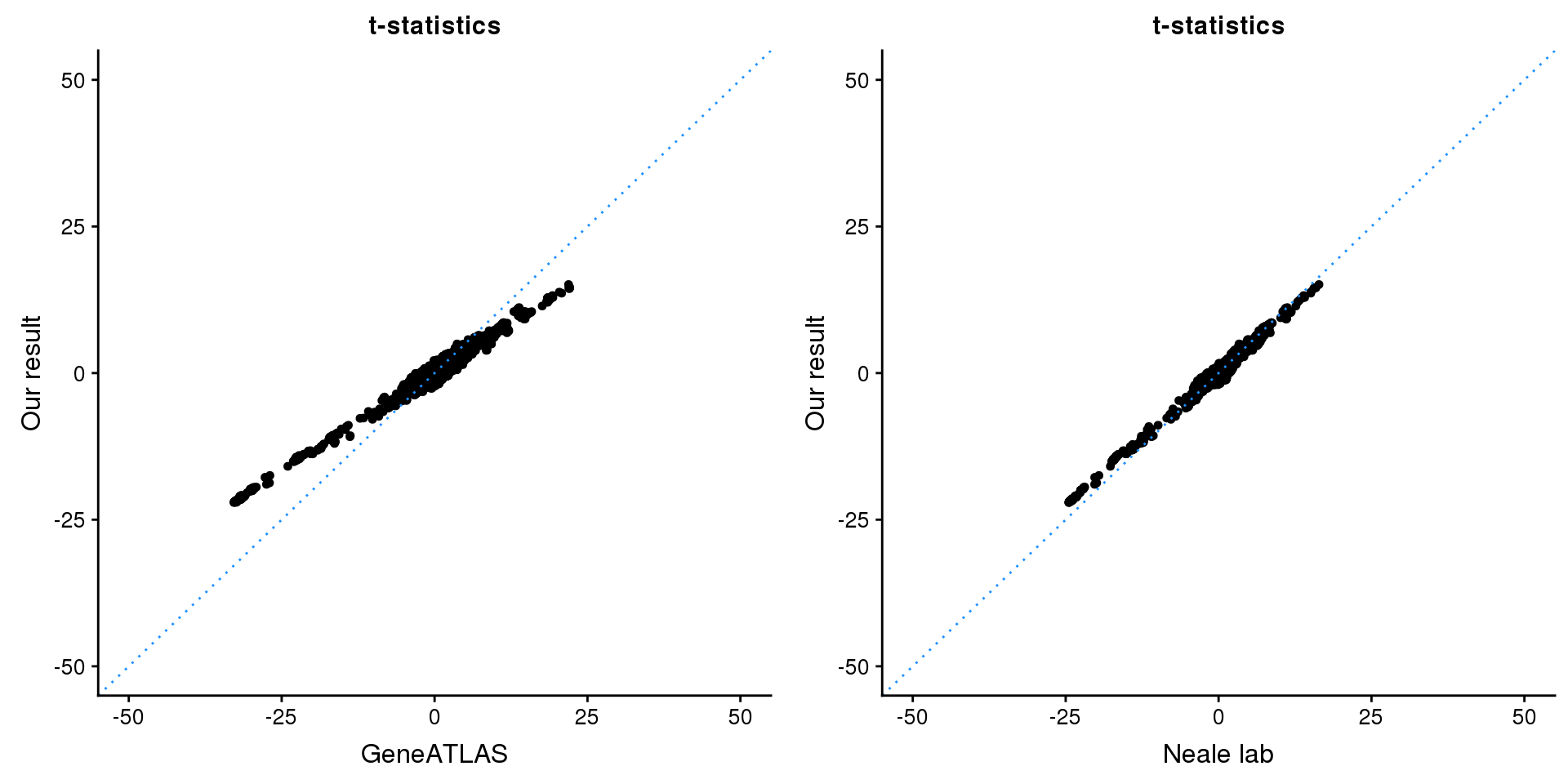
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(3,4,6,15),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)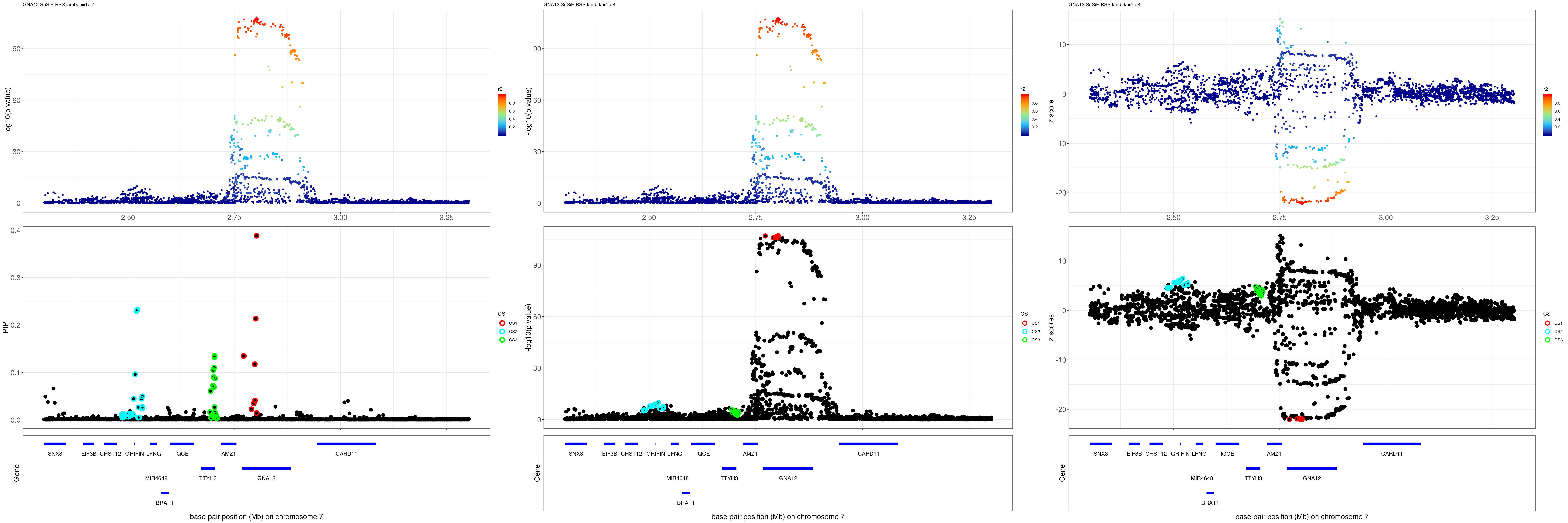
ADAMTS17
Get summary statistics:
region.name = 'ADAMTS17'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "4859 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)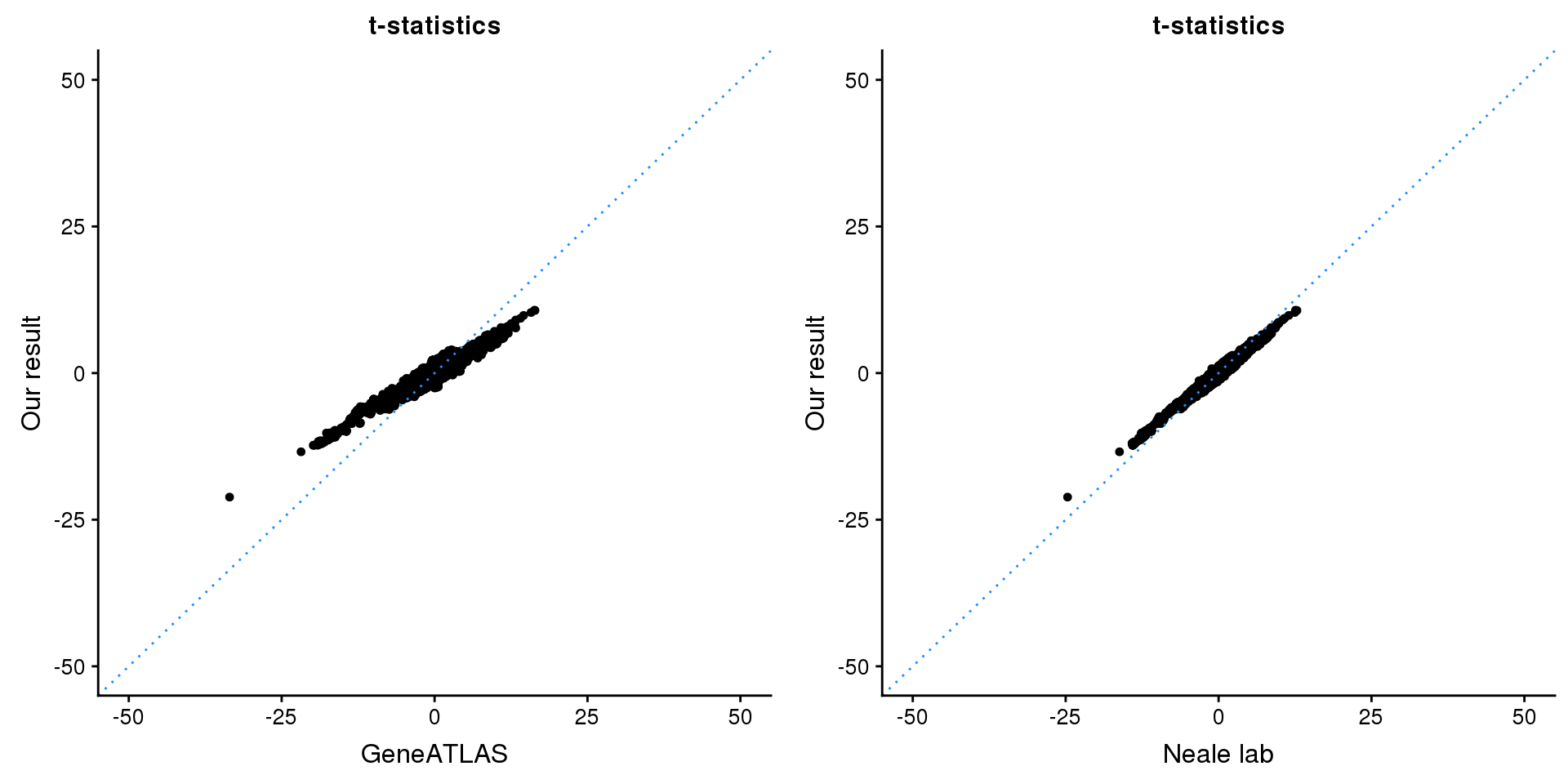
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(4,5,12),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)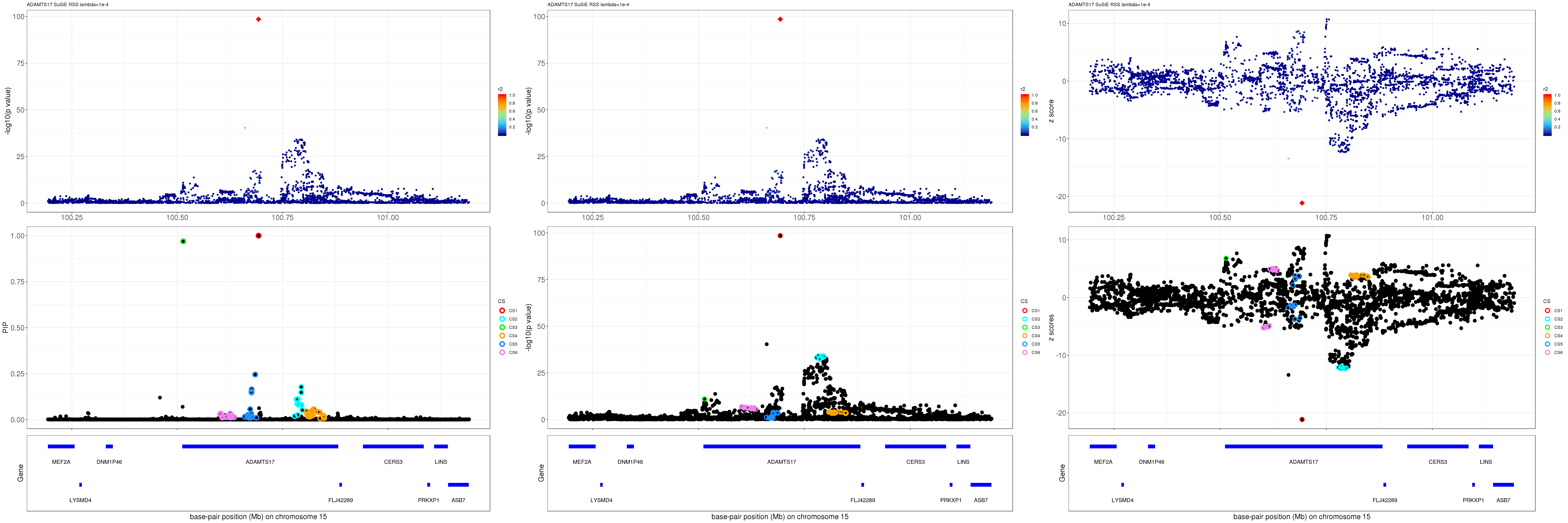
CDK6
Get summary statistics:
region.name = 'CDK6'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "1816 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)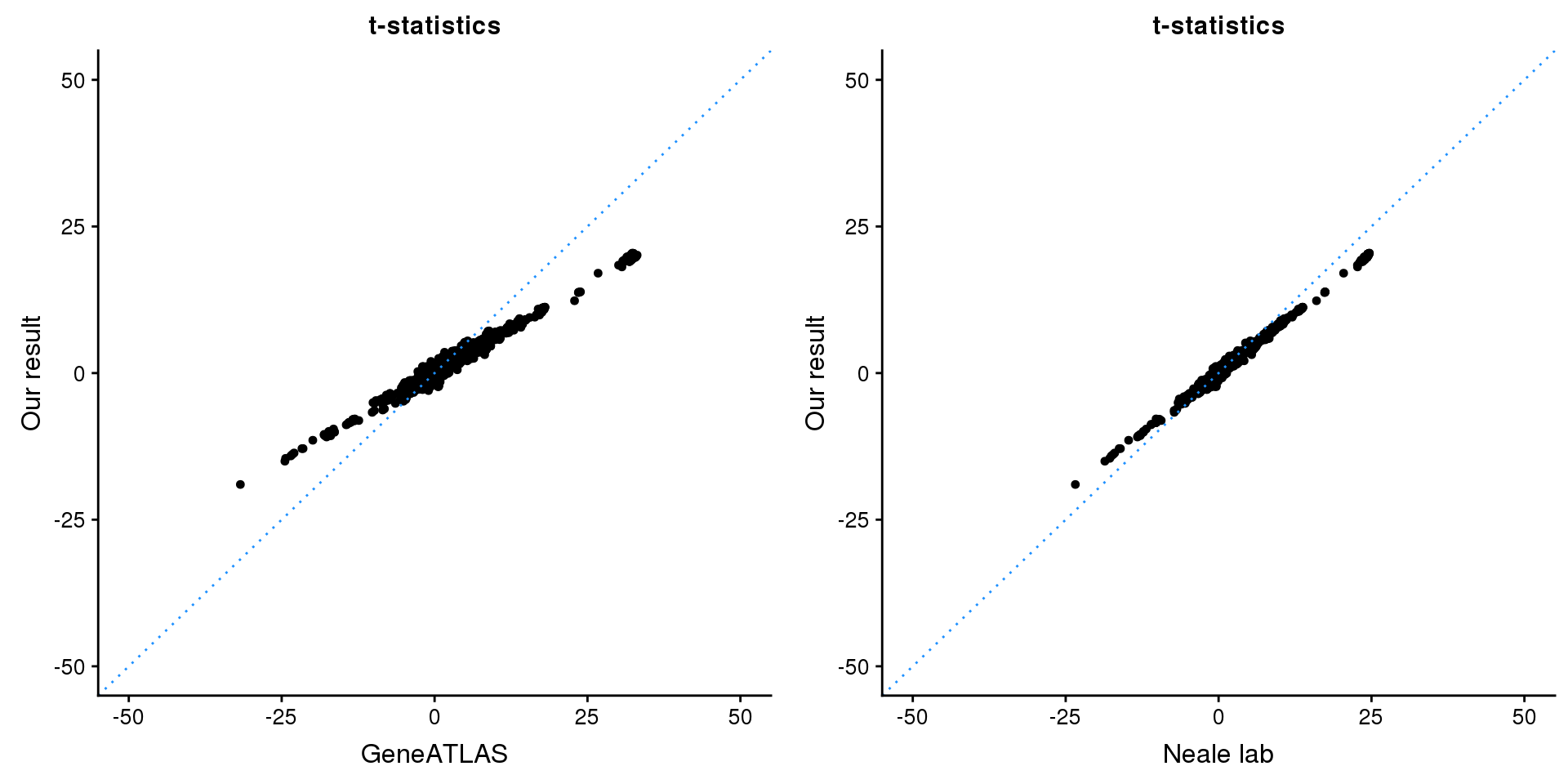
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(2,7,13),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)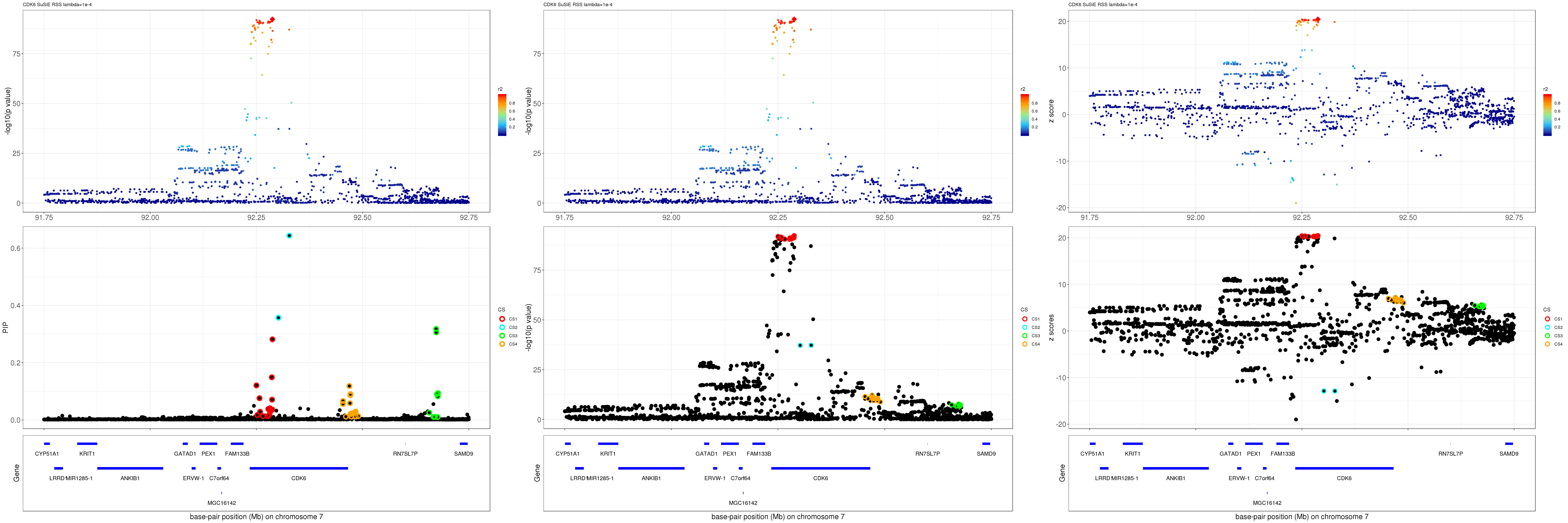
MTMR11
Get summary statistics:
region.name = 'MTMR11'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "1080 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)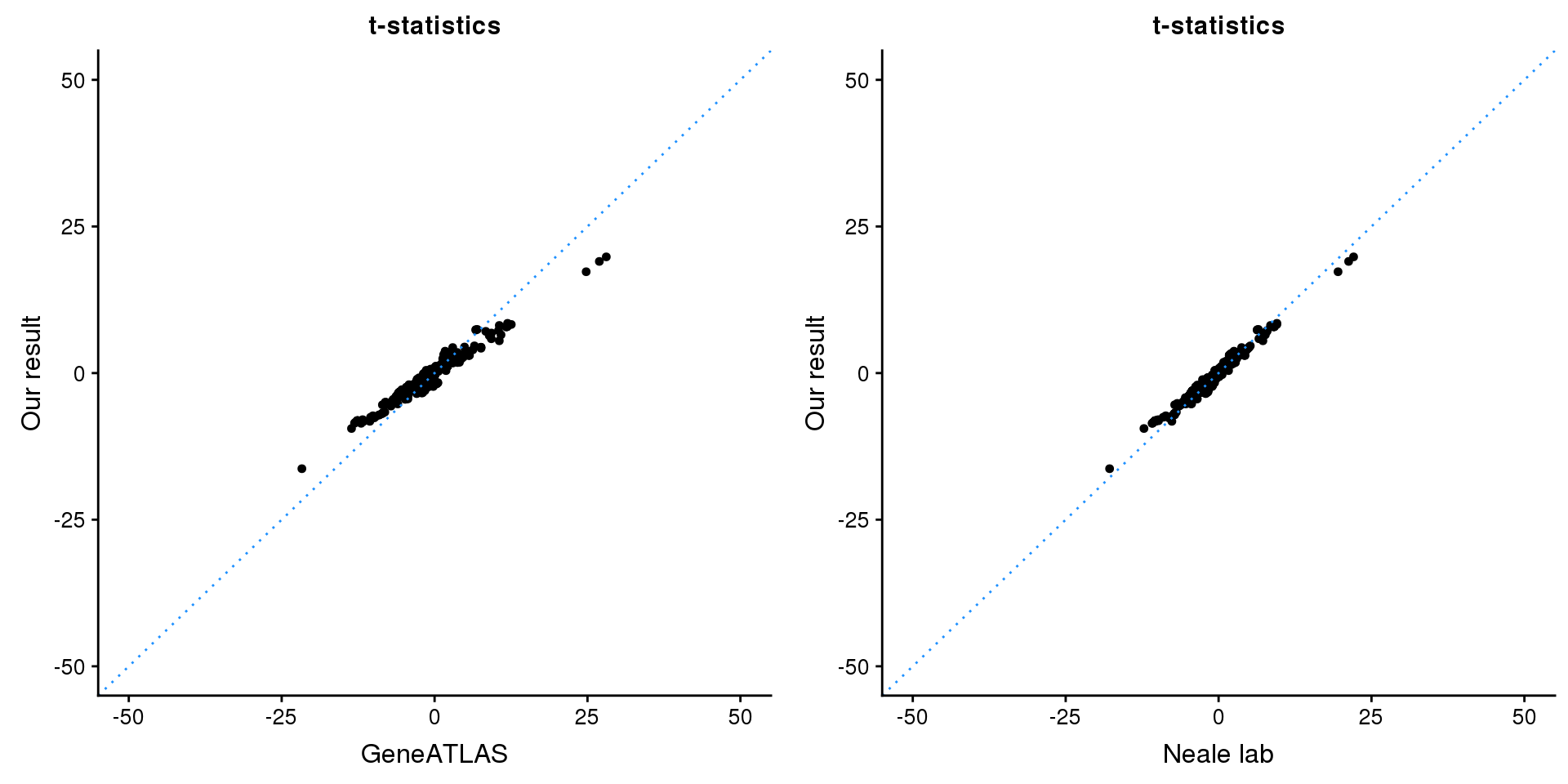
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(1,3,4,8,17,30),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)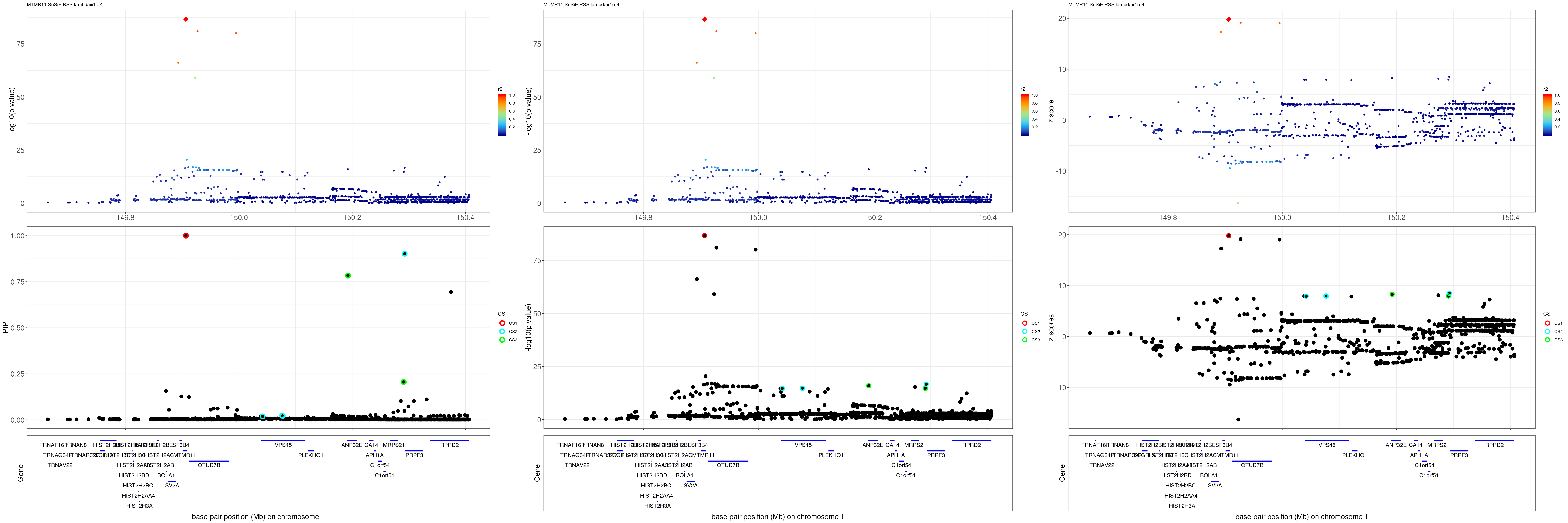
ADAMTSL3
Get summary statistics:
region.name = 'ADAMTSL3'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2259 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)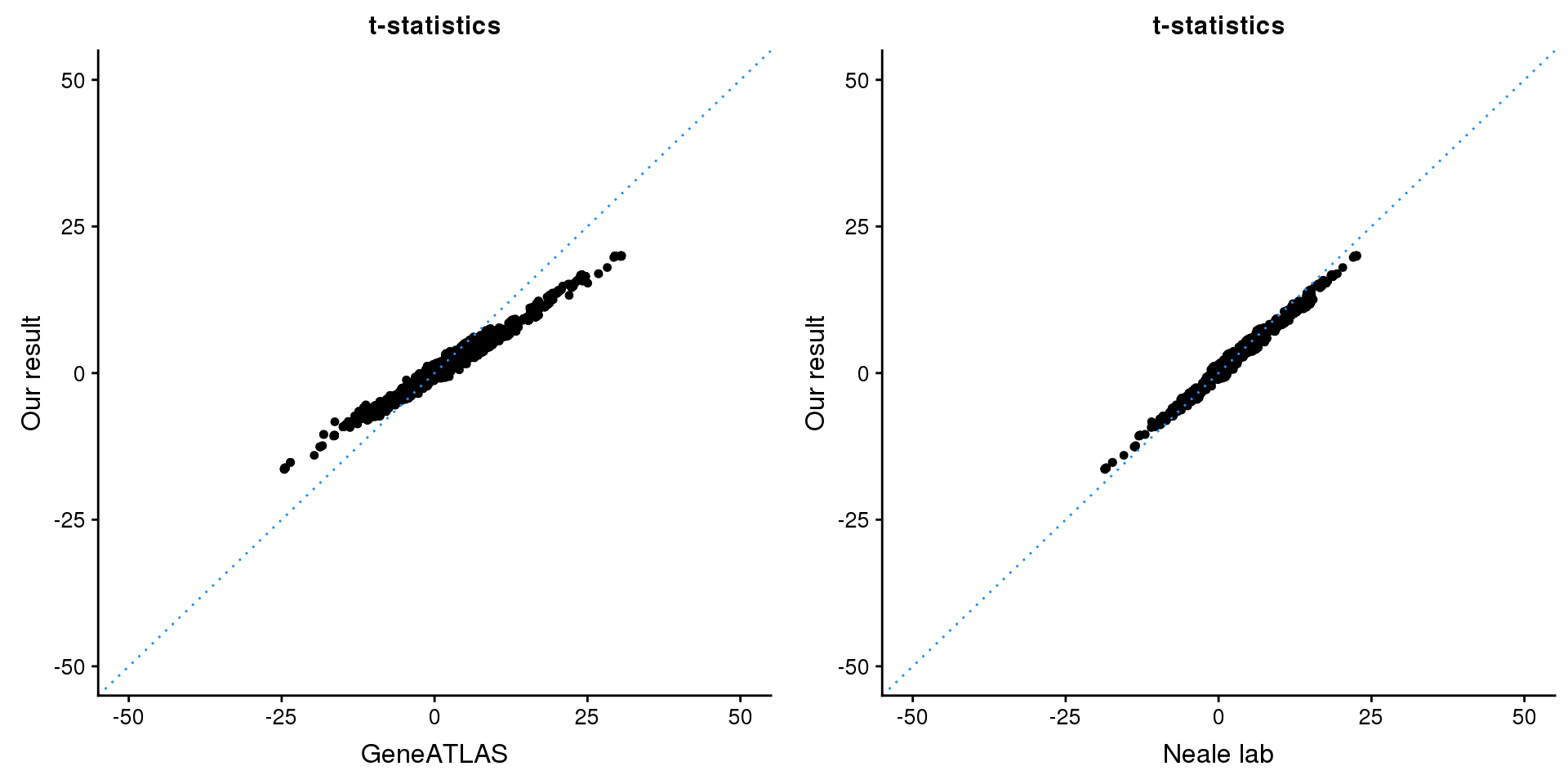
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(3,4,6:12,14,16),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)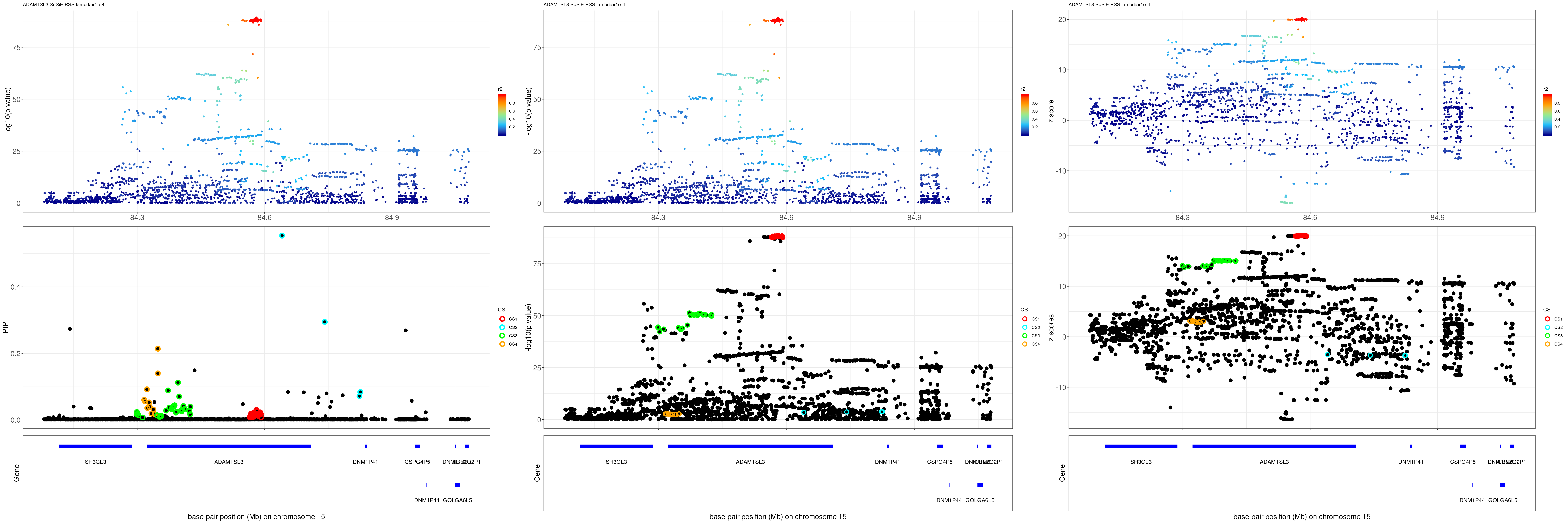
DLEU1
Get summary statistics:
region.name = 'DLEU1'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2500 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)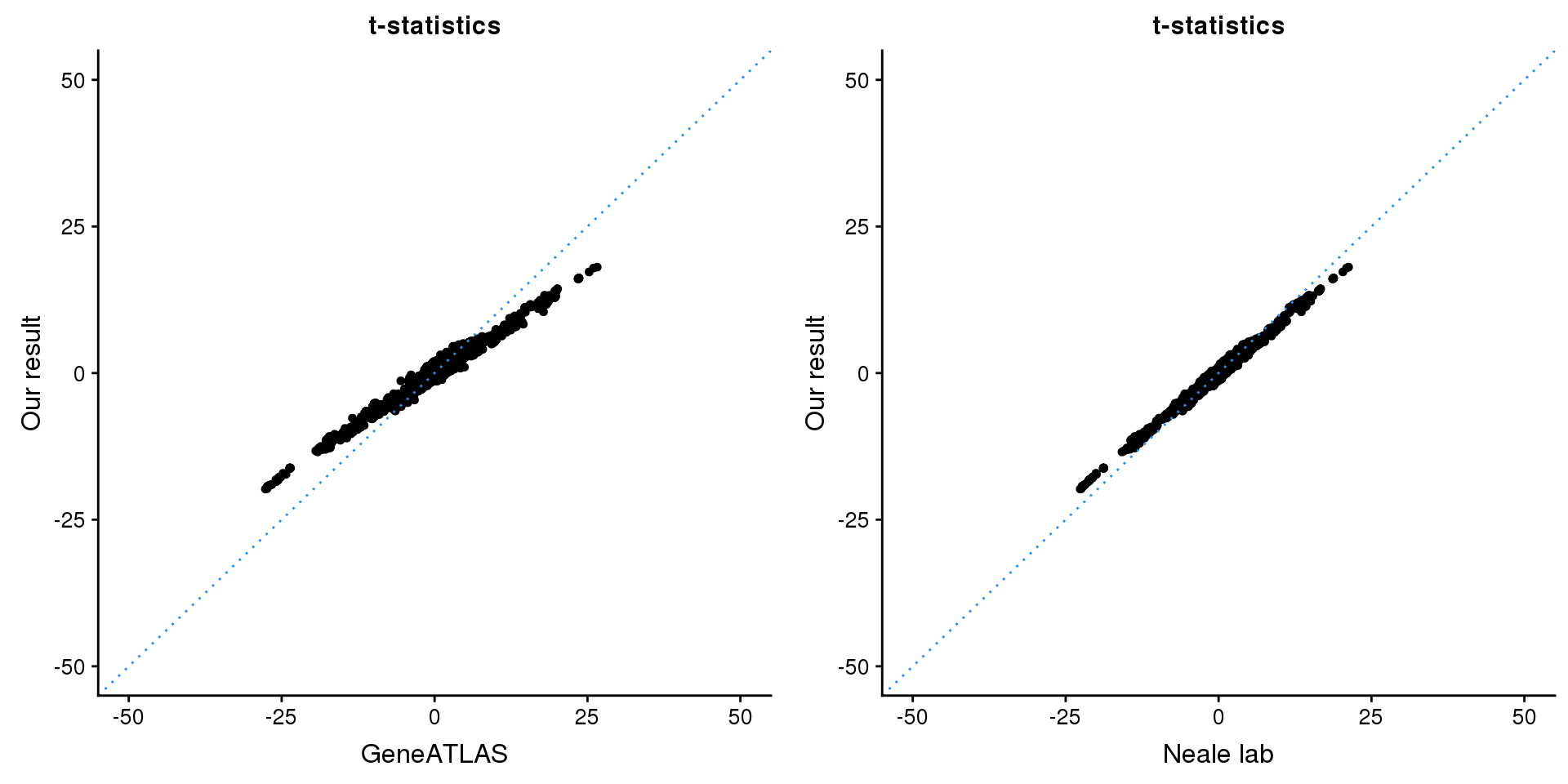
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)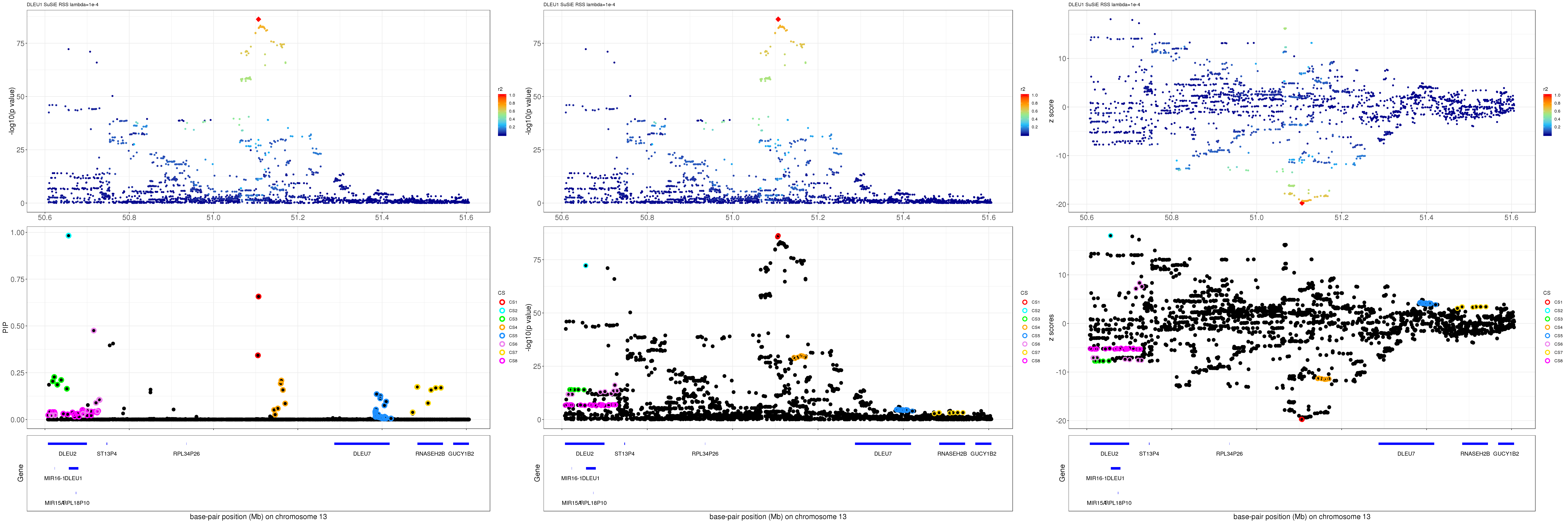
ACAN
Get summary statistics:
region.name = 'ACAN'
file = dir("data/", pattern=paste0("height.",region.name,'.XtX'))
ss.dat = readRDS(paste0('data/', file))
betas = as.vector(ss.dat$Xty/diag(ss.dat$XtX))
rss = c(ss.dat$yty) - betas * ss.dat$Xty
se = as.vector(sqrt(rss/((ss.dat$n-1)*diag(ss.dat$XtX))))
z = betas/se
pval = 2*pnorm(-abs(z))
R = as.matrix(t(ss.dat$XtX * (1/sqrt(diag(ss.dat$XtX)))) * (1/ sqrt(diag(ss.dat$XtX))))
names(z) = rownames(R)
print(paste0(length(z), ' SNPs included'))[1] "2846 SNPs included"Comparing with Neale and GeneATLAS results:
map = fread(paste0('data/region-variants-', region.name, '.csv.gz'))
z.table = data.frame('id' = gsub('_.*$', '', names(z)), 'z' = z)
map = merge(map, z.table, by='id', all = TRUE)
p1 <- ggplot(map,aes(x = NBETA/NSE,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "GeneATLAS",y = "Our result",title = "t-statistics")
p2 <- ggplot(map,aes(x = neale_tstat,y = z)) +
geom_point(shape = 20,size = 2,na.rm = TRUE) +
geom_abline(intercept = 0,slope = 1,color = "dodgerblue",
linetype = "dotted") +
xlim(c(-50,50)) +
ylim(c(-50,50)) +
theme_cowplot(font_size = 12) +
labs(x = "Neale lab",y = "Our result",title = "t-statistics")
gridExtra::grid.arrange(p1, p2, ncol=2)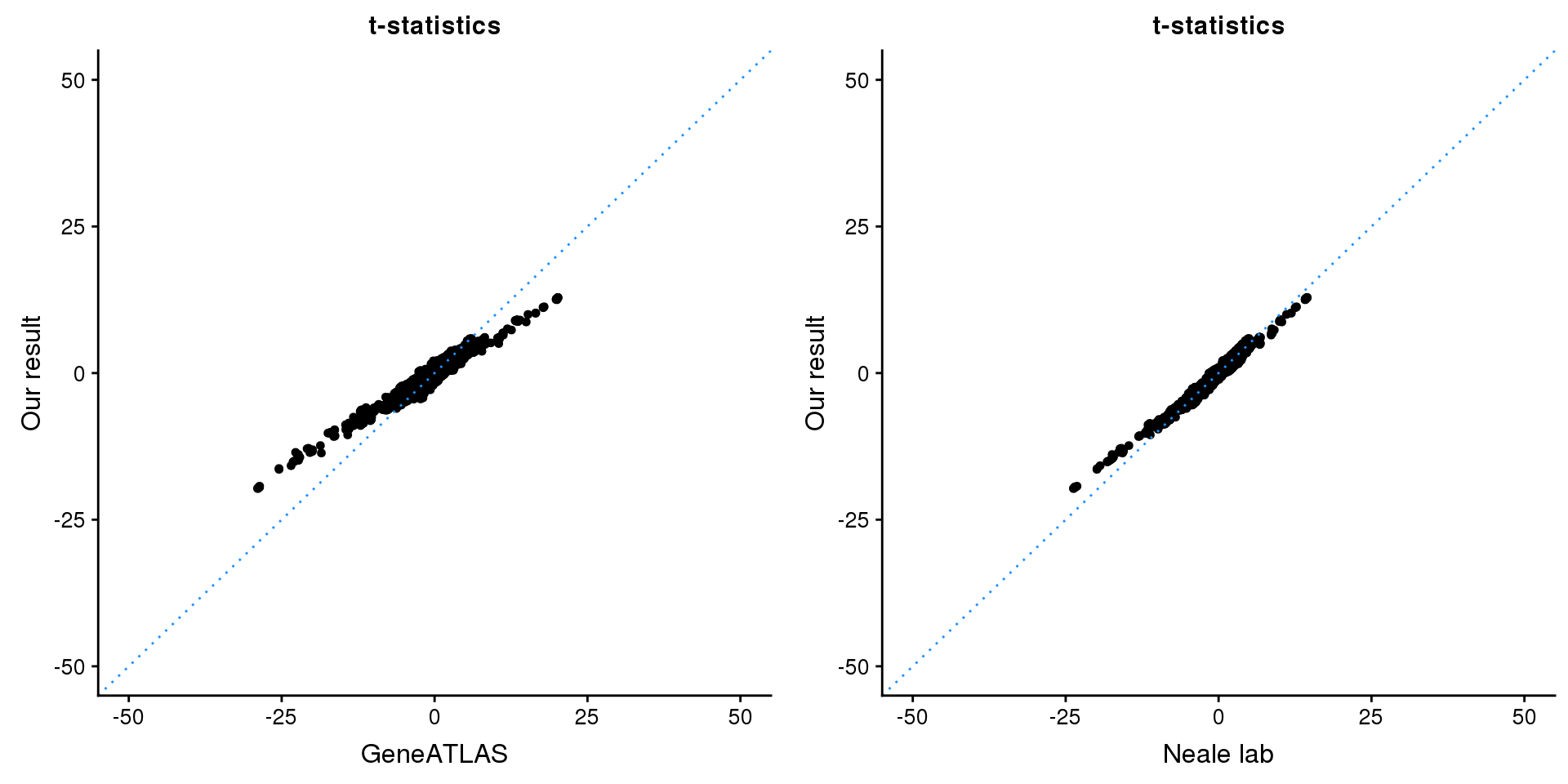
start.pos <- min(ss.dat$pos$POS)
stop.pos <- max(ss.dat$pos$POS)
plot.genes <- subset(genes,
chromosome == unique(ss.dat$pos$`#CHROM`) &
((chr_start > start.pos & chr_start < stop.pos) |
(chr_stop > start.pos & chr_start < stop.pos)) & feature_type == 'GENE')
gene.pos.map = plot.genes %>% select(feature_name, chr_start, chr_stop)
colnames(gene.pos.map) = c('geneName', 'start', 'end')
gene.pos.map = as.data.frame(gene.pos.map)
gene.pos.map = gene.pos.map %>% mutate(start = start/1e6, end = end/1e6)
gene.pos.map = gene.pos.map[-c(13,20),]SuSiE result:
mod_rss = susie_rss(z = z, R = R, lambda=1e-04)Warning in susie_rss(z = z, R = R, lambda = 1e-04): This method is under
active development, so it should not be considered stable.Warning in susie_rss(z = z, R = R, lambda = 1e-04): The maximum number of
non-zero effects is greater than 1, this feature is experimental.z.max = which.max(abs(z))
pip_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'))
p_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'p')
z_rss = susie_plot_locuszoom(z, mod_rss, pos = ss.dat$pos$POS/1e6, chr = unique(ss.dat$pos$`#CHROM`), gene.pos.map = gene.pos.map, ld = R[z.max,], z.ref.name = names(z.max), title = paste0(region.name,' SuSiE RSS lambda=1e-4'), y.susie = 'z')
grid.arrange(pip_rss, p_rss, z_rss, ncol=3)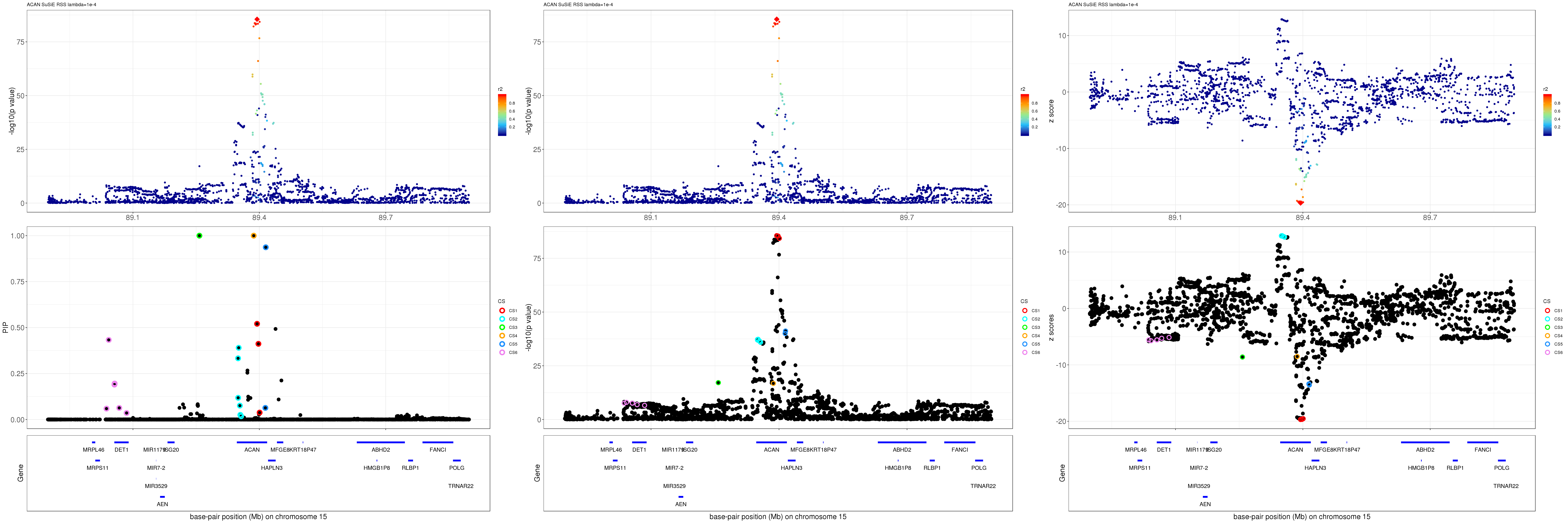
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cowplot_0.9.4 readr_1.3.1 gridExtra_2.3 Matrix_1.2-15
[5] data.table_1.12.0 susieR_0.8.1.0545 ggplot2_3.1.1 dplyr_0.8.0.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.1 pillar_1.3.1 compiler_3.5.1
[4] later_0.7.5 git2r_0.26.1 plyr_1.8.4
[7] workflowr_1.5.0 R.methodsS3_1.7.1 R.utils_2.7.0
[10] tools_3.5.1 digest_0.6.18 evaluate_0.12
[13] tibble_2.0.1 gtable_0.2.0 lattice_0.20-38
[16] egg_0.4.5 pkgconfig_2.0.2 rlang_0.3.1
[19] yaml_2.2.0 withr_2.1.2 stringr_1.3.1
[22] knitr_1.20 hms_0.4.2 fs_1.3.1
[25] rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5
[28] glue_1.3.0 R6_2.3.0 rmarkdown_1.10
[31] purrr_0.3.2 magrittr_1.5 whisker_0.3-2
[34] backports_1.1.2 scales_1.0.0 promises_1.0.1
[37] htmltools_0.3.6 assertthat_0.2.1 colorspace_1.4-0
[40] httpuv_1.4.5 labeling_0.3 stringi_1.2.4
[43] lazyeval_0.2.1 munsell_0.5.0 crayon_1.3.4
[46] R.oo_1.22.0