Simple simulations for a simplified version of web-boost-2
Last updated: 2020-07-16
Checks: 5 2
Knit directory: causal-TWAS/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20191103) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/causalTWAS/causal-TWAS/code/fit_mr.ash.R | code/fit_mr.ash.R |
| ~/causalTWAS/causal-TWAS/code/mr.ash2.R | code/mr.ash2.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/.ipynb_checkpoints/
Ignored: data/
Untracked files:
Untracked: analysis/figure/
Untracked: mr.ash2s.bk
Untracked: mr.ash2s.lasso-beta.Rd
Untracked: mr.ash2s.rds
Unstaged changes:
Modified: README.md
Modified: analysis/simulation_simpleveb-boost2.Rmd
Modified: code/fit_mr.ash.R
Modified: code/gen_mr.ash2_output.R
Modified: code/input_reformat.R
Modified: code/mr.ash2.R
Modified: code/run_gwas_snp.R
Modified: code/run_simulate_data.R
Modified: code/run_test_mr.ash2s.R
Modified: code/workflow-ashtest3.ipynb
Modified: code/workflow-data.ipynb
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | ca1d8bc | simingz | 2020-07-02 | mr.ash2s lasso beta init |
| html | ca1d8bc | simingz | 2020-07-02 | mr.ash2s lasso beta init |
| html | 3a24c5e | simingz | 2020-06-11 | plotly |
| Rmd | 8f01adb | simingz | 2020-05-28 | mr.ash2s |
| html | 8f01adb | simingz | 2020-05-28 | mr.ash2s |
Simulation of data
20 blocks:
- Each block has either gene or SNP effect
- Each block has 99 SNPs and 1 gene. Each gene is linear sum of the previous 3 SNPs.
- Each block, either the gene or the last eQTL has non-zero effect on trait
The first 4 blocks have gene effect.
set.seed(1)
N <- 4000
nblocks <- 20
block.size <- 100
p <- nblocks * block.size
n.eQTL <- 3 # number of eQTLs per gene
sigma.eQTL <- 0.5 # eQTL effect size
sigma.SNP <- 0.1 # effect size of causal SNP on trait
sigma.gene <- 0.1 # effect size of causal gene on trait
X <- matrix(rep(0,0), nrow=N, ncol=0)
gamma.gene <- rep(0, nblocks) # indicator of genes
gamma.gene[1:4] <- 1
beta <- numeric(0)
SNP.idx <- numeric(0)
for (i in 1:nblocks) {
# sample SNP data
X.block.SNP <- matrix(rnorm(N*(block.size-1)), nrow=N, ncol=block.size-1)
SNP.idx <- c(SNP.idx, 1:(block.size-1) + (i-1)*block.size)
# generate gene data: use the previous few SNPs as eQTL
effects.eQTL <- rnorm(n.eQTL, 0, sigma.eQTL)
X.block.gene <- X.block.SNP[, (block.size - n.eQTL):(block.size - 1)] %*% effects.eQTL
X.block = cbind(X.block.SNP, X.block.gene)
X <- cbind(X, X.block)
# sample beta
if (gamma.gene[i] == 1) { # gene effect in this block
beta.SNP <- rep(0, block.size - 1)
beta.gene <- rnorm(1, 0, sigma.gene)
} else { # SNP effect in this block
beta.SNP <- c(rep(0, block.size - 2), rnorm(1, 0, sigma.SNP))
beta.gene <- 0
}
beta.block <- c(beta.SNP, beta.gene)
beta <- c(beta, beta.block)
}
sigma.e <- 1
y <- X %*% beta + rnorm(N, 0, sigma.e)
gene.idx <- (1:nblocks) * block.sizeRun mr.ash
summary_mr.ash <- function(fit){
cat("pi1 = ", 1-fit$pi[[1]], "\n")
pve <- get_pve(fit)
cat("pve : ", pve, "\n")
}
plot_beta <- function(beta,beta.pm, ...){
plot( beta, pch=19, col ="darkgreen", ...)
points(beta.pm, pch =19, col = "red")
legend("topright", legend=c("true beta", "posterior mean"),
col=c("darkgreen", "red"), pch=19)
}fit <- mr.ash(X, y, method="caisa")
summary_mr.ash(fit)pi1 = 0.0114093
pve : 0.1476845 plot_beta(beta[gene.idx], fit$beta[gene.idx], main = "beta for gene effect")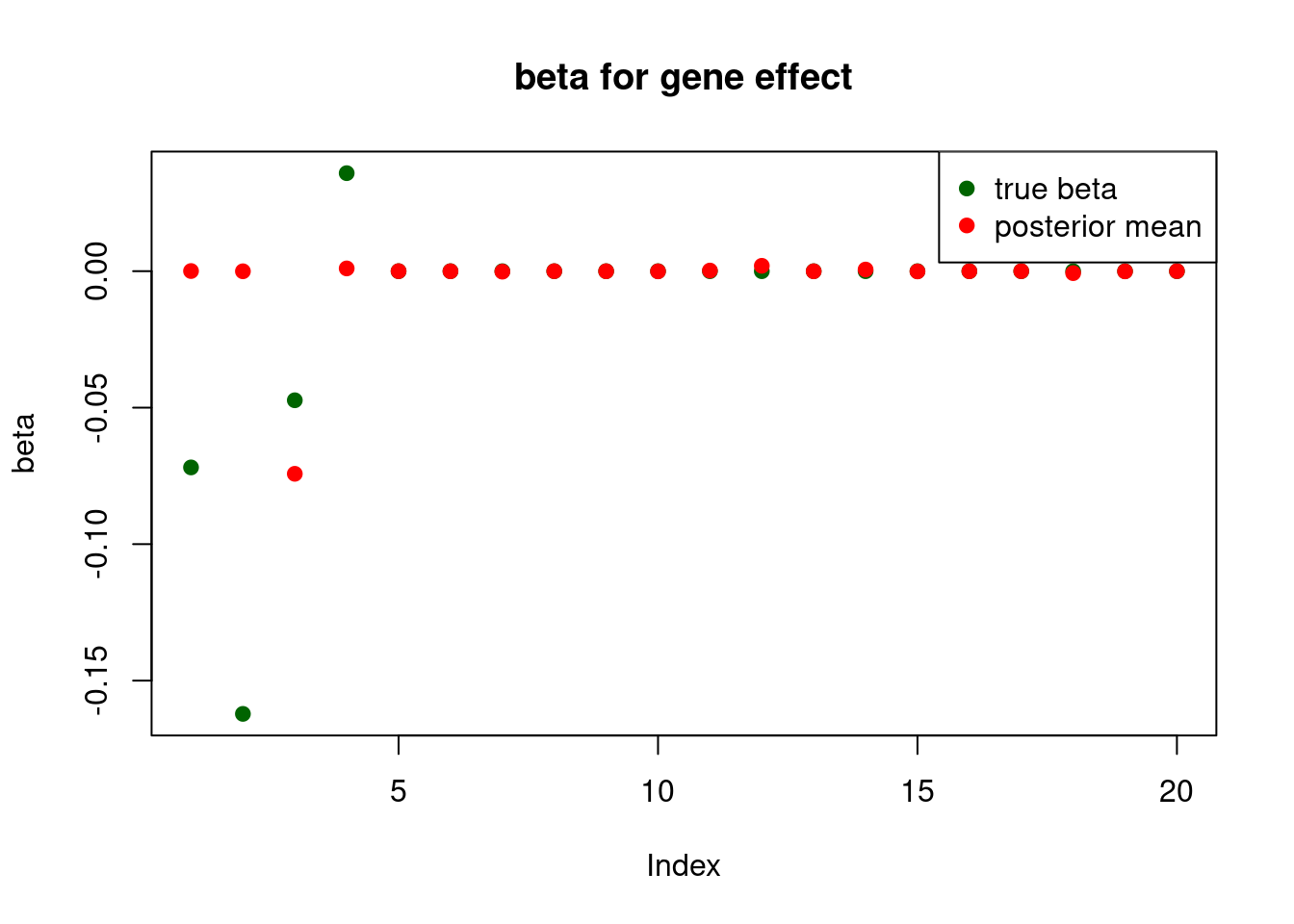
plot_beta(beta[SNP.idx], fit$beta[SNP.idx], main = "beta for SNP effect")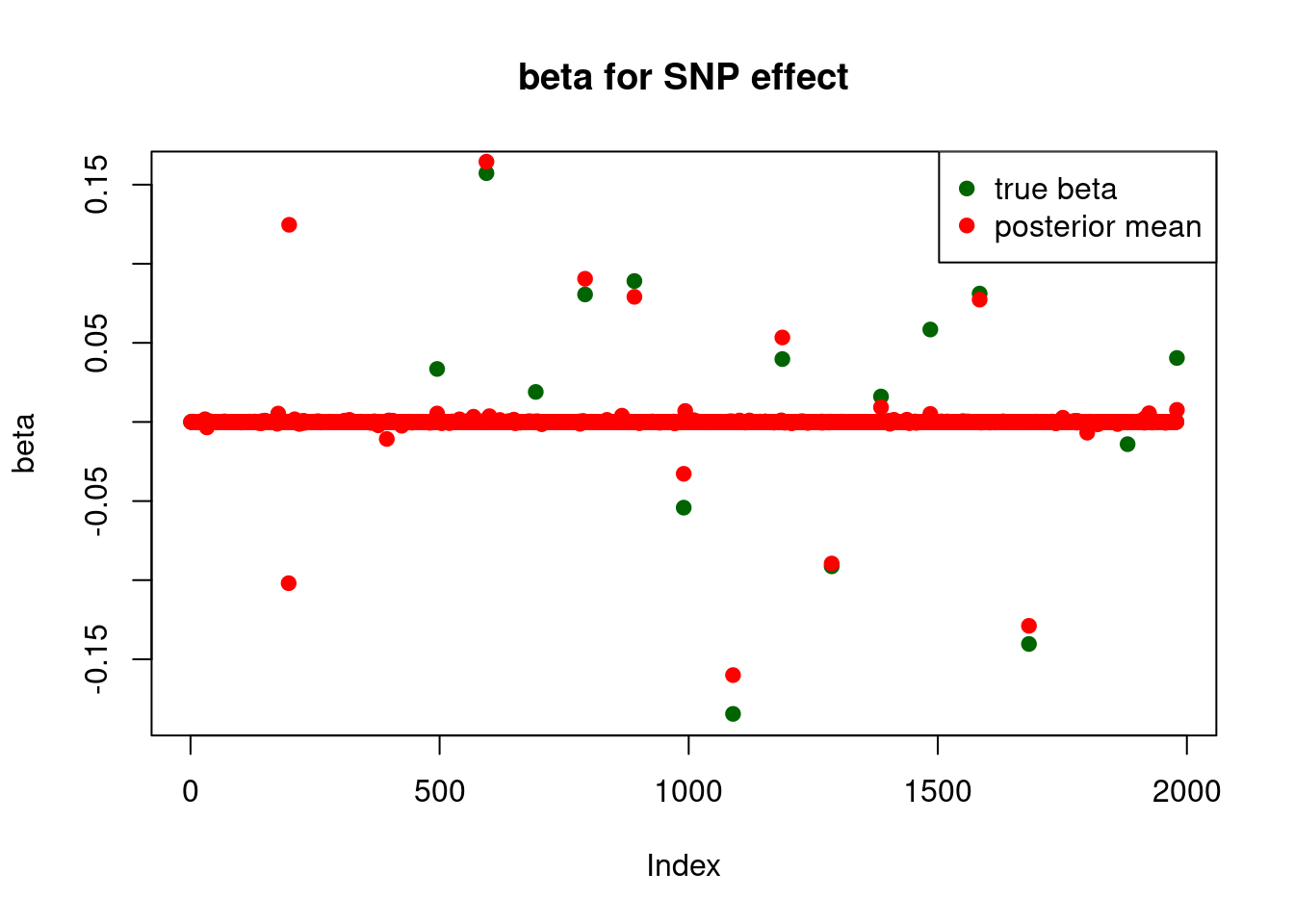
Run a simplified version of veb-boost (mr.ash2s)
start with gene
X.gene <- X[, gene.idx]
X.SNP <- as_FBM(X[, SNP.idx])
fit <- mr.ash2s( X.SNP, X.gene, y, init.order = "expr-snp")Warning in if (mr.ash.init == "lasso") {: the condition has length > 1 and
only the first element will be usedprint("for gene effect: ")[1] "for gene effect: "summary_mr.ash(fit$fit2)pi1 = 0.007331657
pve : 0.1237024 plot_beta(beta[gene.idx], fit$fit2$beta, main = "beta for gene effect")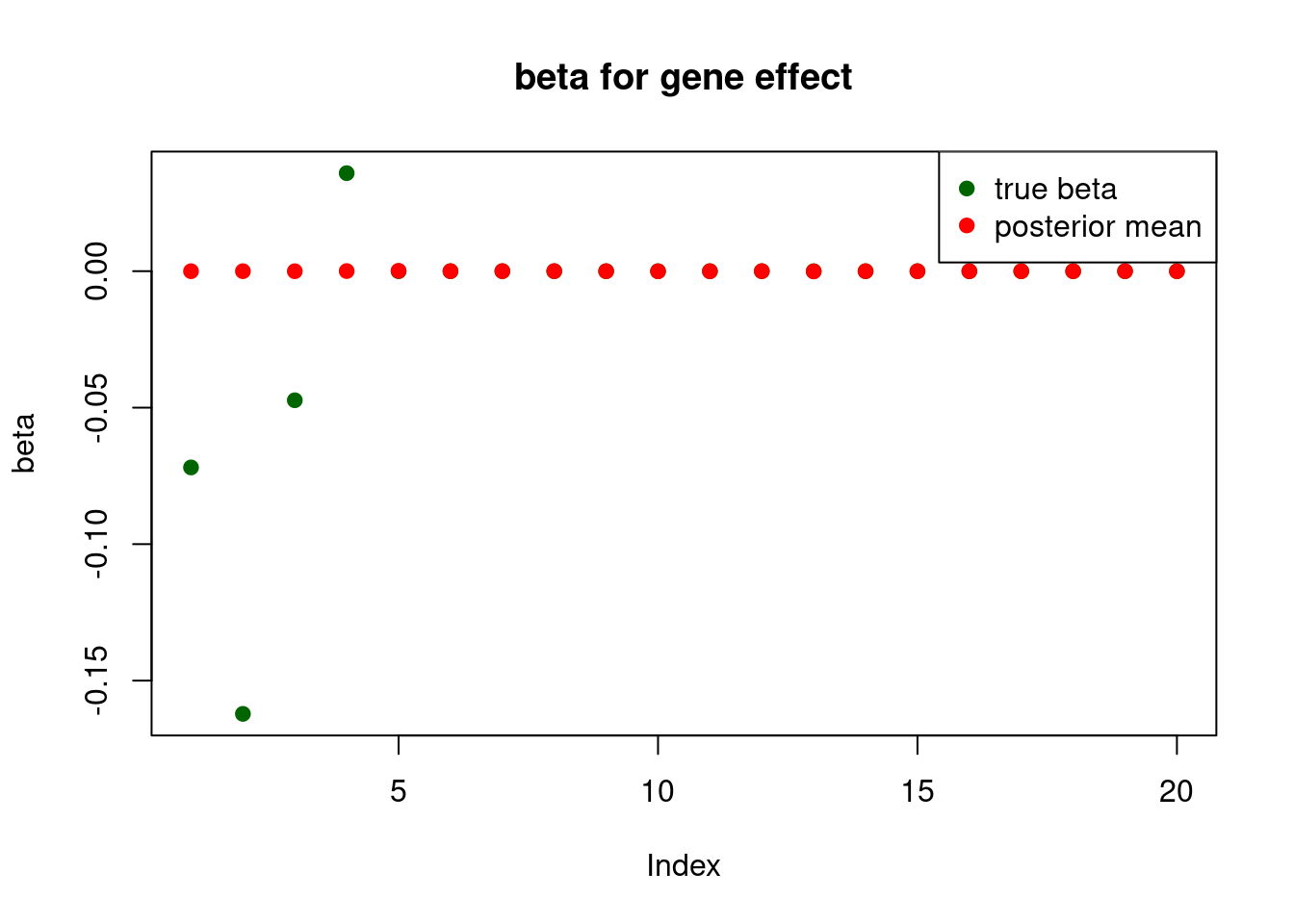
print("for SNP effect: ")[1] "for SNP effect: "summary_mr.ash(fit$fit1)pi1 = 0.4053821
pve : 0.03753964 plot_beta(beta[SNP.idx], fit$fit1$beta, main = "beta for SNP effect")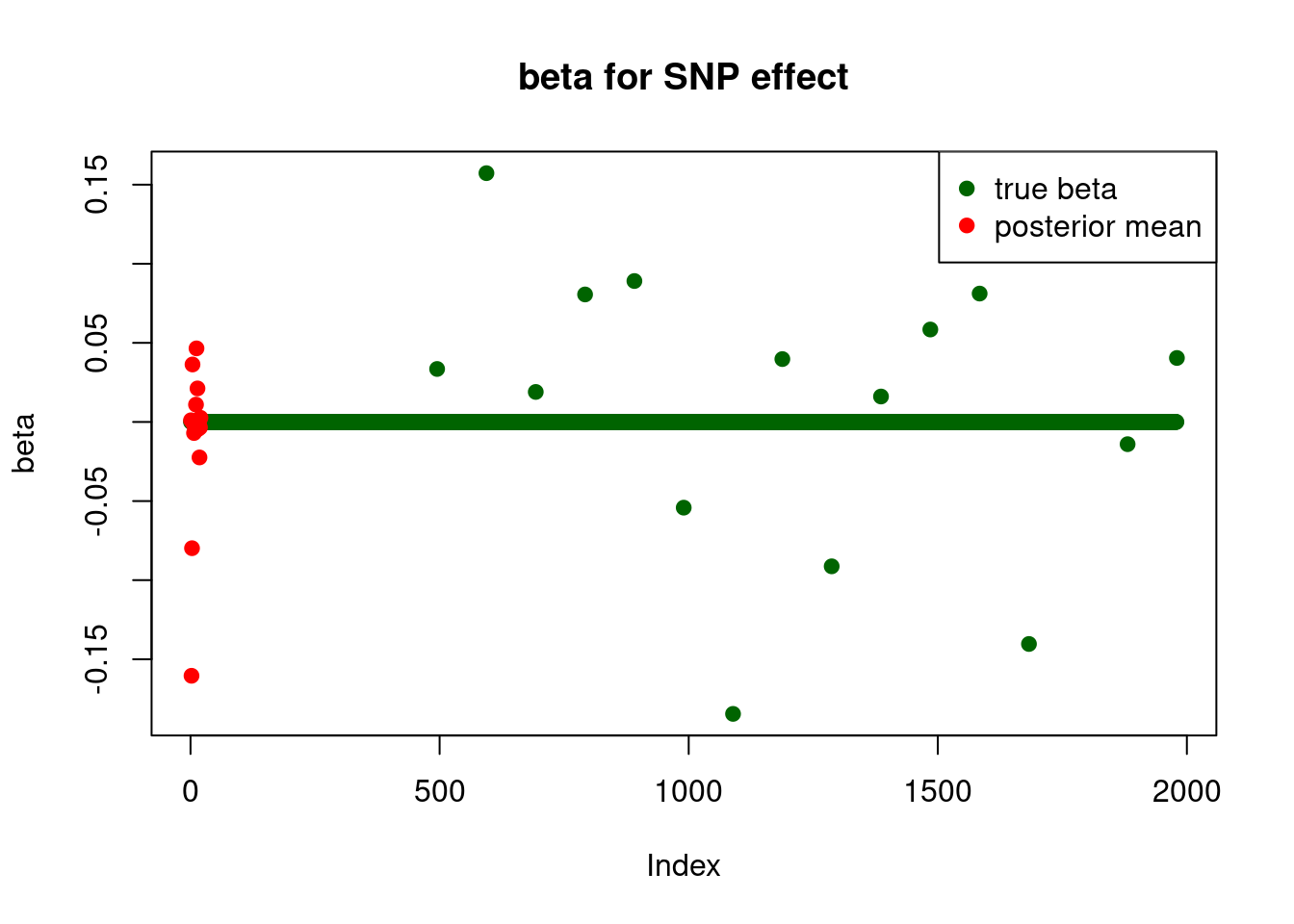
start with SNP
fit <- mr.ash2s(X.SNP, X.gene, y, init.order = "snp-expr")Warning in if (mr.ash.init == "lasso") {: the condition has length > 1 and
only the first element will be usedprint("for gene effect: ")[1] "for gene effect: "summary_mr.ash(fit$fit2)pi1 = 0.007331658
pve : 0.1237024 plot_beta(beta[gene.idx], fit$fit2$beta, main = "beta for gene effect")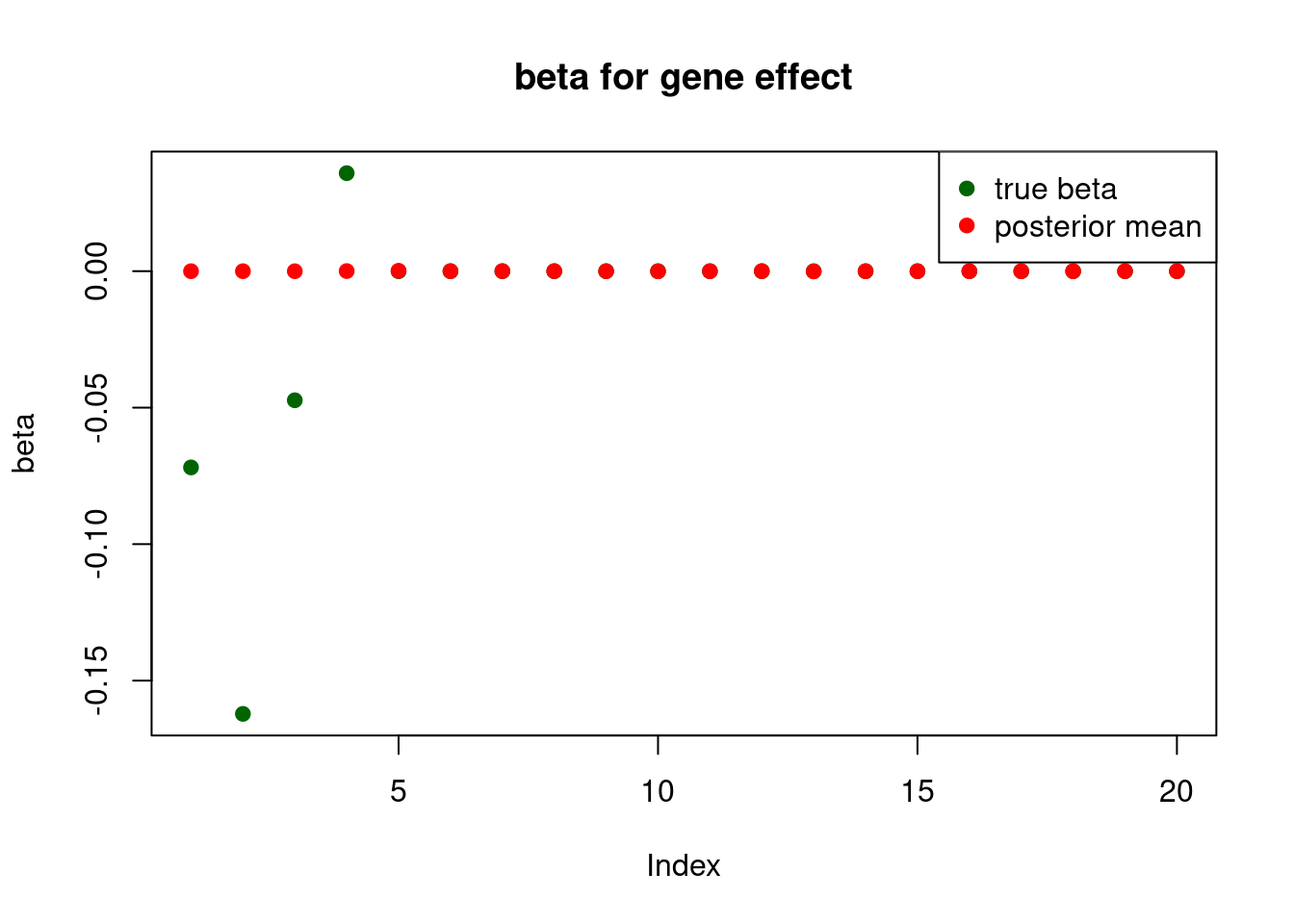
print("for SNP effect: ")[1] "for SNP effect: "summary_mr.ash(fit$fit1)pi1 = 0.405382
pve : 0.03753965 plot_beta(beta[SNP.idx], fit$fit1$beta, main = "beta for SNP effect")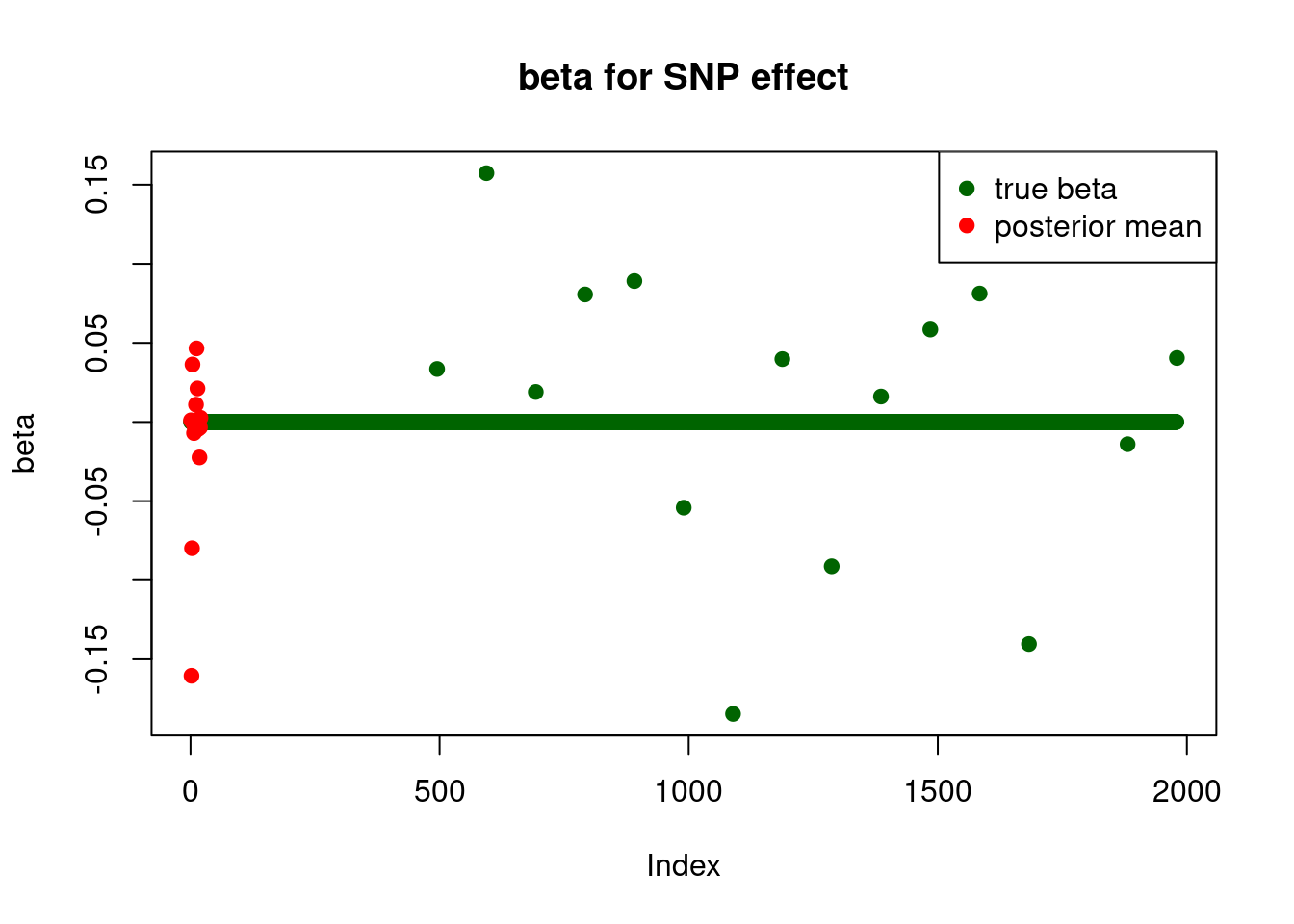
init with lasso
fit <- mr.ash2s( X.SNP,X.gene, y, mr.ash.init = "lasso")Warning in if (init.order == "expr-snp") {: the condition has length > 1
and only the first element will be usedprint("for gene effect: ")[1] "for gene effect: "summary_mr.ash(fit$fit2)pi1 = 0.007331657
pve : 0.1237024 plot_beta(beta[gene.idx], fit$fit2$beta, main = "beta for gene effect")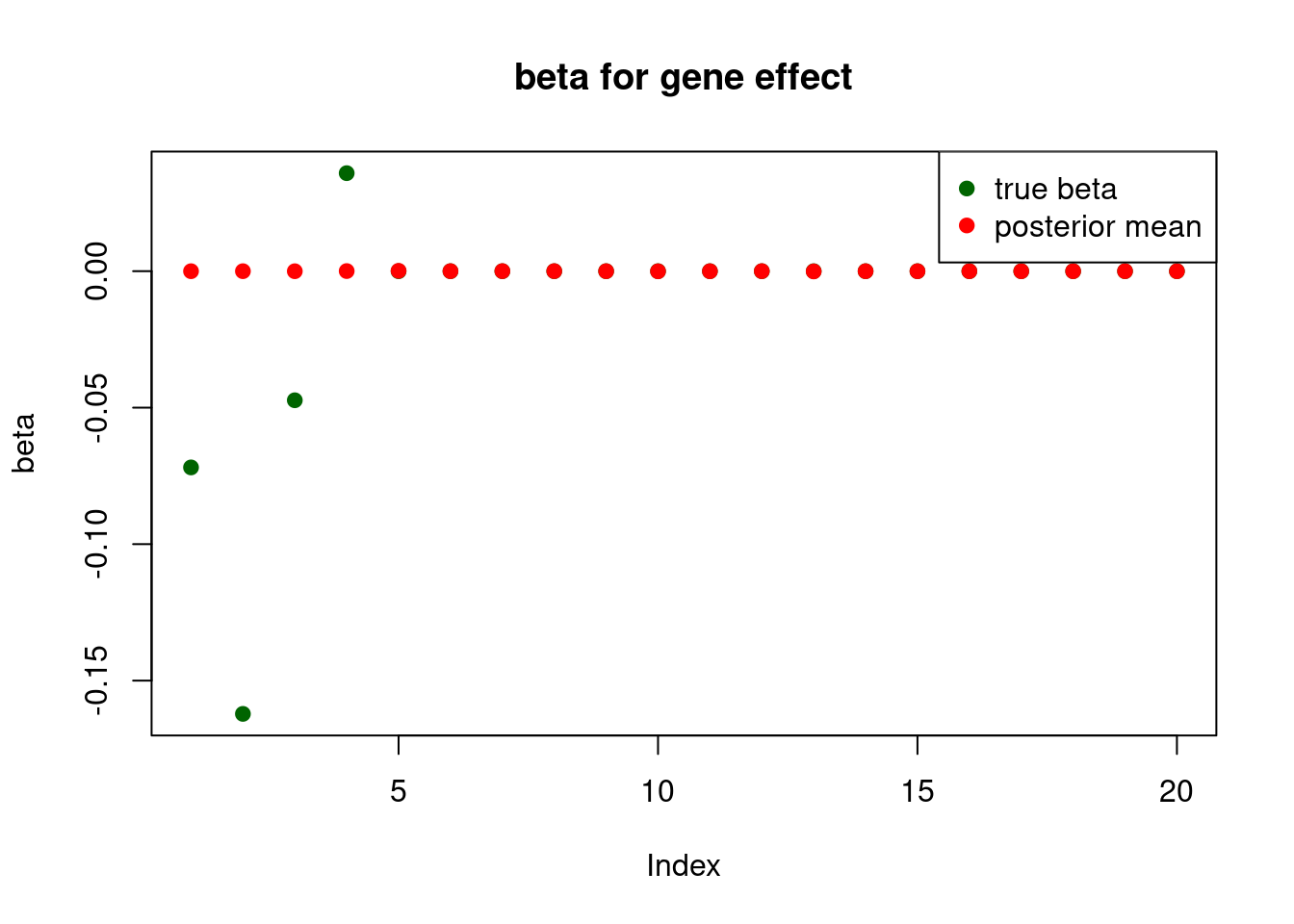
| Version | Author | Date |
|---|---|---|
| ca1d8bc | simingz | 2020-07-02 |
print("for SNP effect: ")[1] "for SNP effect: "summary_mr.ash(fit$fit1)pi1 = 0.4053821
pve : 0.03753964 plot_beta(beta[SNP.idx], fit$fit1$beta, main = "beta for SNP effect")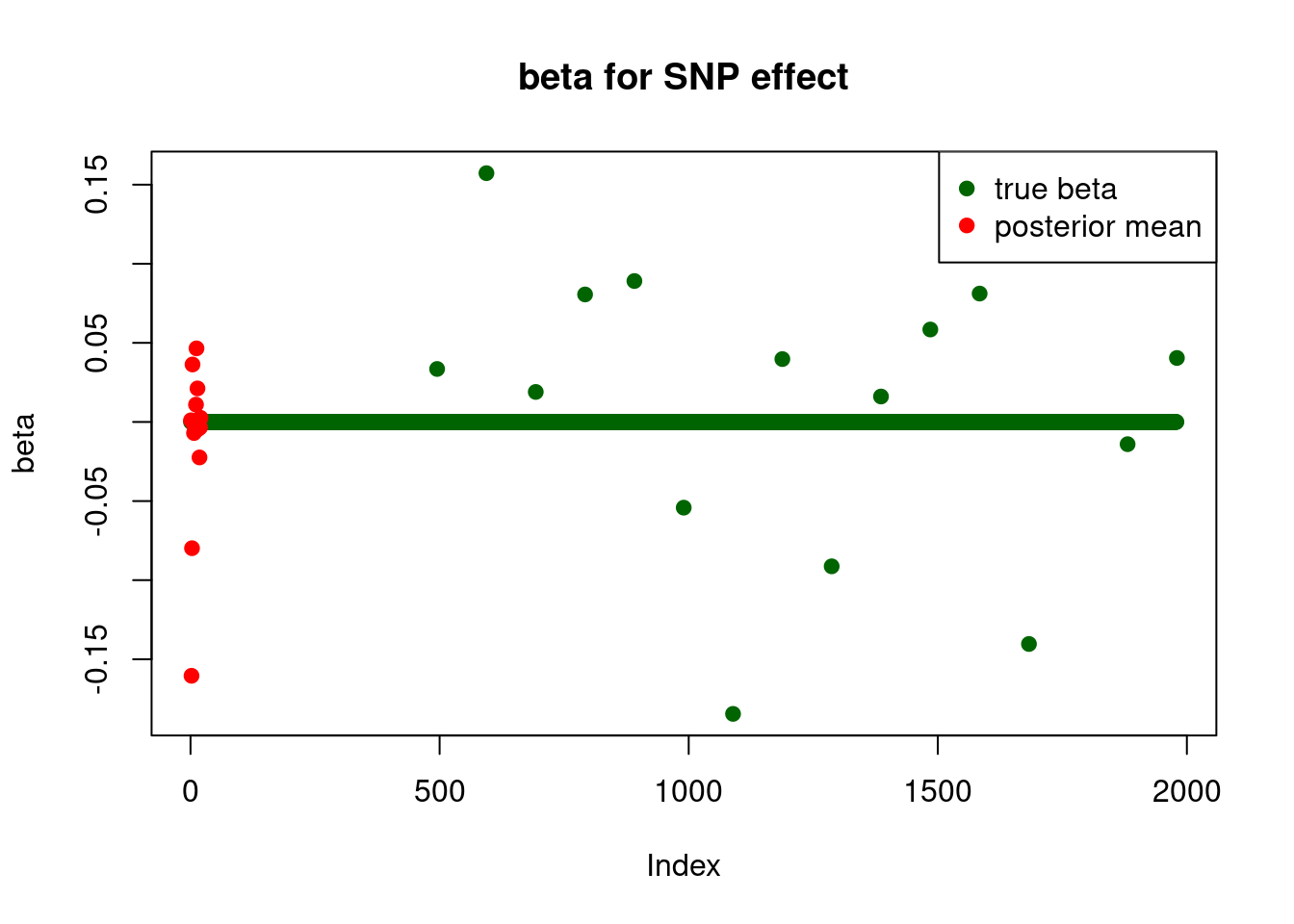
| Version | Author | Date |
|---|---|---|
| ca1d8bc | simingz | 2020-07-02 |
init with lasso SNP
fit <- mr.ash2s(X.SNP, X.gene, y, mr.ash.init = "lassoSNP")Warning in if (init.order == "expr-snp") {: the condition has length > 1
and only the first element will be usedprint("for gene effect: ")[1] "for gene effect: "summary_mr.ash(fit$fit2)pi1 = 0.007331662
pve : 0.1237026 plot_beta(beta[gene.idx], fit$fit2$beta, main = "beta for gene effect")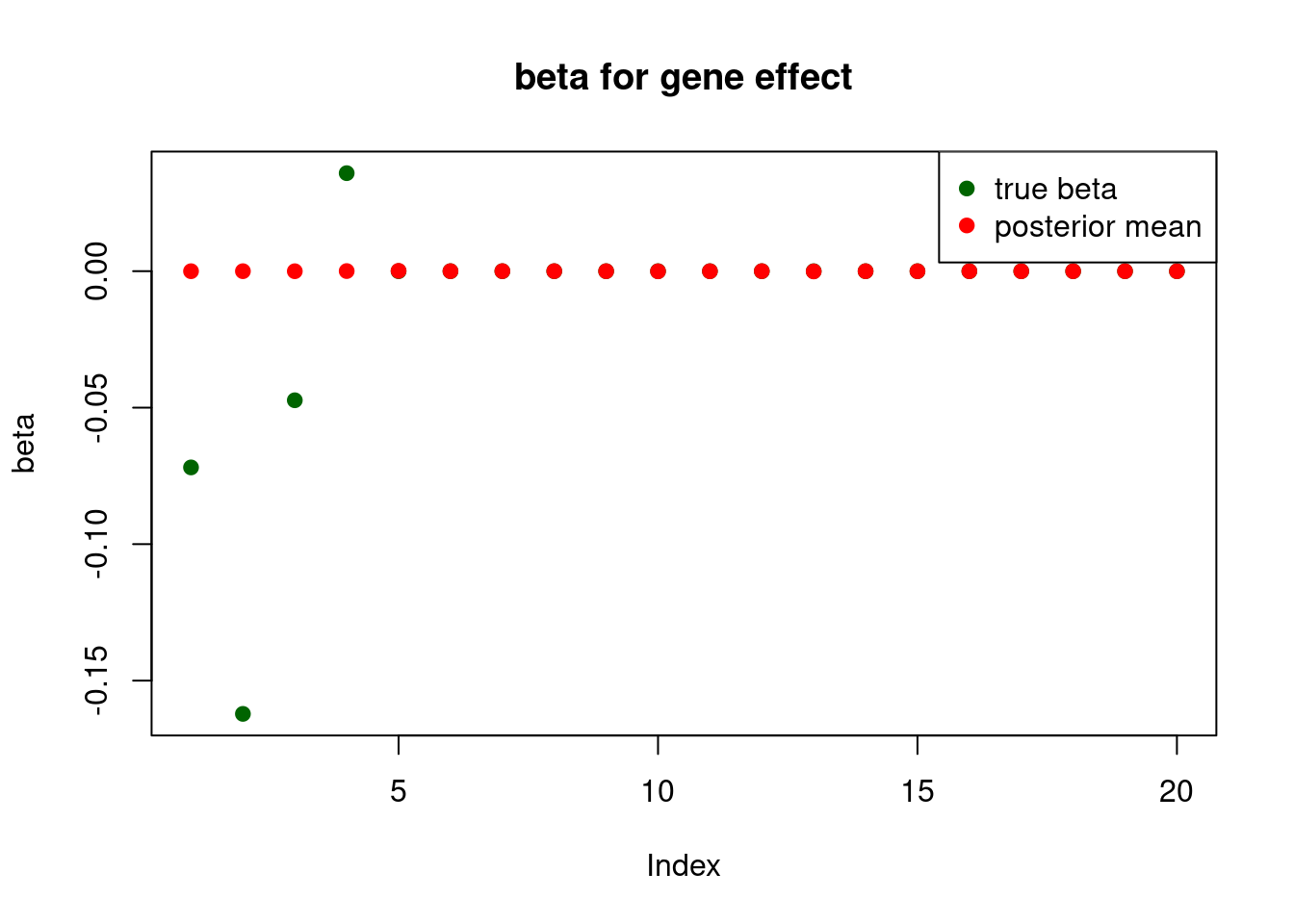
| Version | Author | Date |
|---|---|---|
| ca1d8bc | simingz | 2020-07-02 |
print("for SNP effect: ")[1] "for SNP effect: "summary_mr.ash(fit$fit1)pi1 = 0.4054002
pve : 0.03753852 plot_beta(beta[SNP.idx], fit$fit1$beta, main = "beta for SNP effect")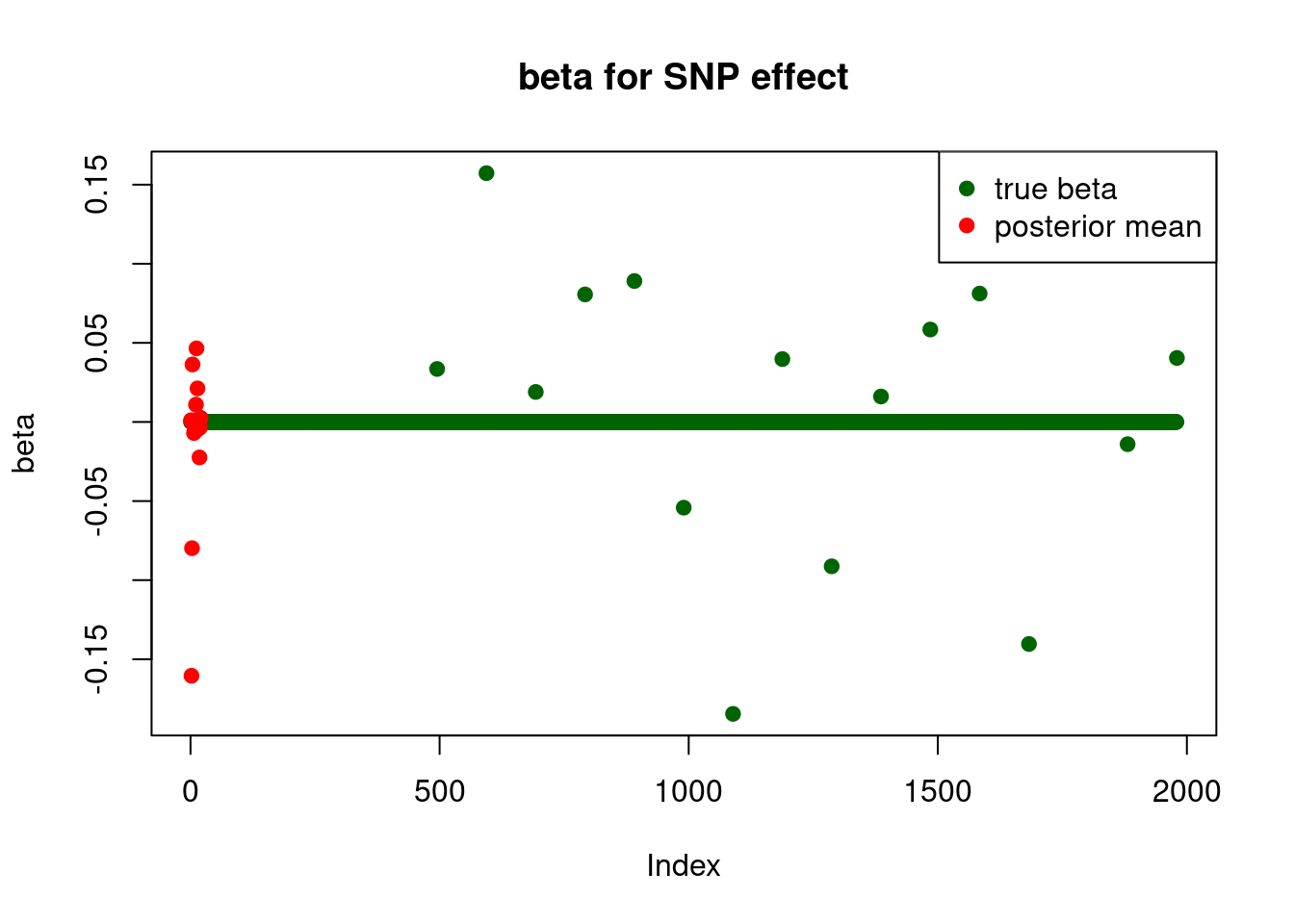
| Version | Author | Date |
|---|---|---|
| ca1d8bc | simingz | 2020-07-02 |
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bigstatsr_1.2.3 biglasso_1.3-7 ncvreg_3.11.2
[4] Matrix_1.2-15 bigmemory_4.5.36 mr.ash.alpha_0.1-34
loaded via a namespace (and not attached):
[1] tidyselect_1.1.0 purrr_0.3.4 lattice_0.20-38
[4] bigassertr_0.1.3 colorspace_1.3-2 vctrs_0.3.1
[7] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0
[10] rlang_0.4.6 later_0.7.5 pillar_1.4.4
[13] glue_1.4.1 foreach_1.4.4 lifecycle_0.2.0
[16] stringr_1.4.0 munsell_0.5.0 gtable_0.2.0
[19] workflowr_1.6.0 codetools_0.2-15 evaluate_0.12
[22] knitr_1.20 doParallel_1.0.15 httpuv_1.4.5
[25] parallel_3.5.1 highr_0.7 Rcpp_1.0.4.6
[28] promises_1.0.1 scales_1.0.0 backports_1.1.2
[31] fs_1.3.1 ggplot2_3.3.1 digest_0.6.25
[34] stringi_1.3.1 bigparallelr_0.2.3 dplyr_1.0.0
[37] grid_3.5.1 rprojroot_1.3-2 cowplot_0.9.4
[40] tools_3.5.1 magrittr_1.5 tibble_3.0.1
[43] crayon_1.3.4 whisker_0.3-2 bigmemory.sri_0.1.3
[46] pkgconfig_2.0.2 ellipsis_0.3.1 rmarkdown_1.10
[49] iterators_1.0.10 R6_2.3.0 flock_0.7
[52] git2r_0.26.1 compiler_3.5.1